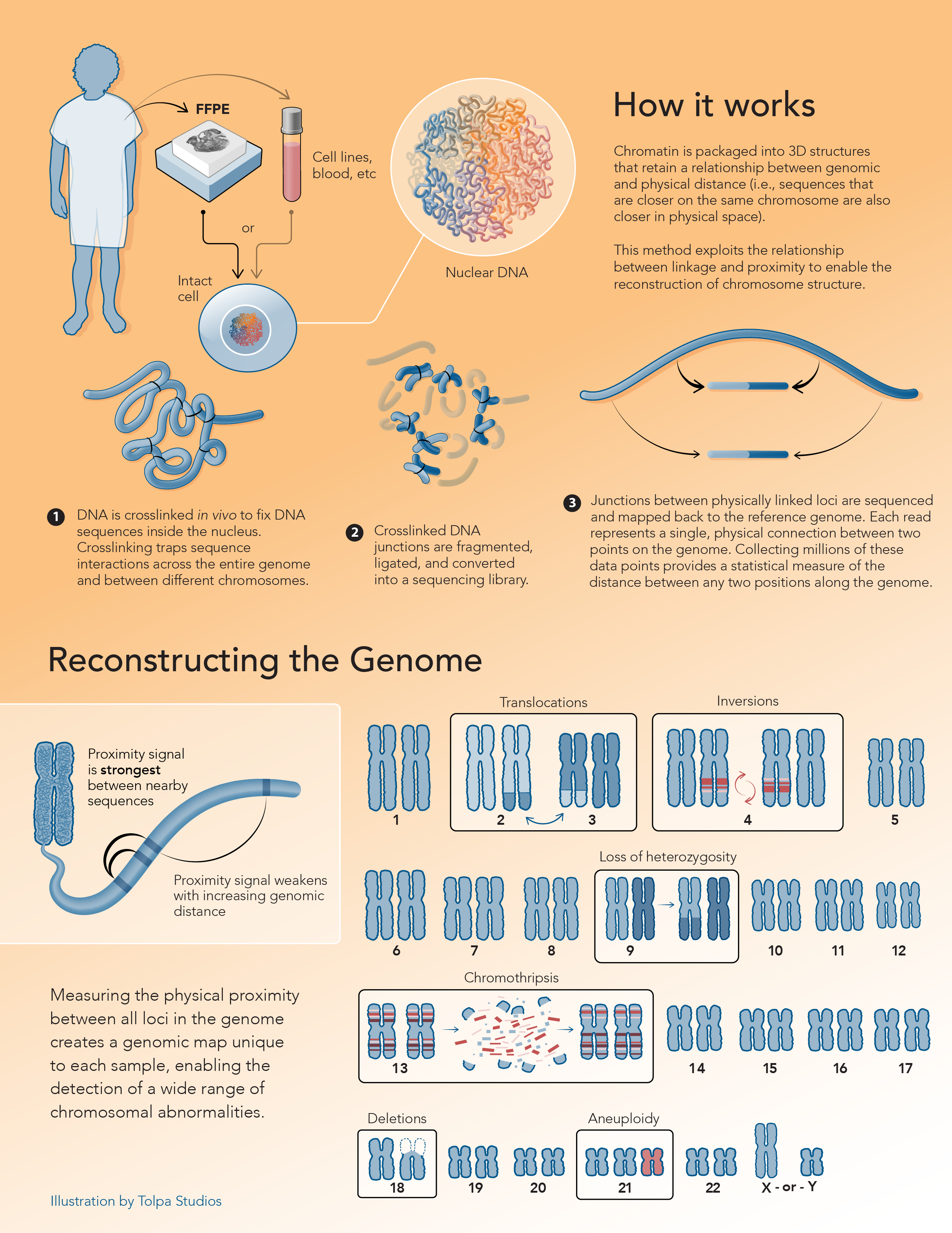
Ultra-long-range Genome Sequencing Technology
High-throughput chromosome conformation capture
Proximity ligation methods connect sequence and structure
Hi-C is one of a suite of chromosome conformation capture (or proximity ligation) techniques originally devised to study the spatial organization of chromatin.1,2 Hi-C employs cost-effective, high-throughput, short-read sequencing to identify the interactions between genomic loci that are co-located in three-dimensional space, but may be separated by significant distances in the linear genome. This powerful methodology has since enabled significant improvements in genome assembly (of humans and other species), as well as structural variant and epigenetic analysis, and has unlocked many applications in metagenomics, microbiology, cytogenomics and oncology.
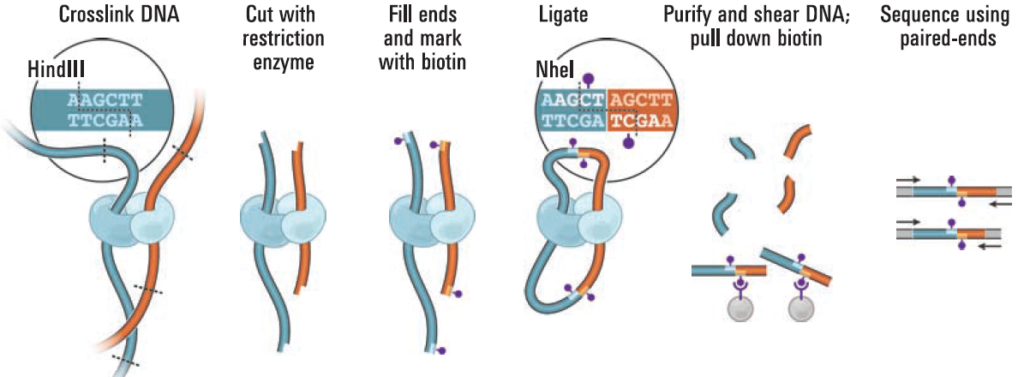
Overview of proximity ligation (Hi-C). From: Lieberman-Aiden E, Dekker J. Science. 2009; 326:289.
Learn more about conformational capture methods:
- Find out what 3C, 4C, 5C, Hi-C and other 3C methods have in common, and how they differ to enable different molecular and structural measurements
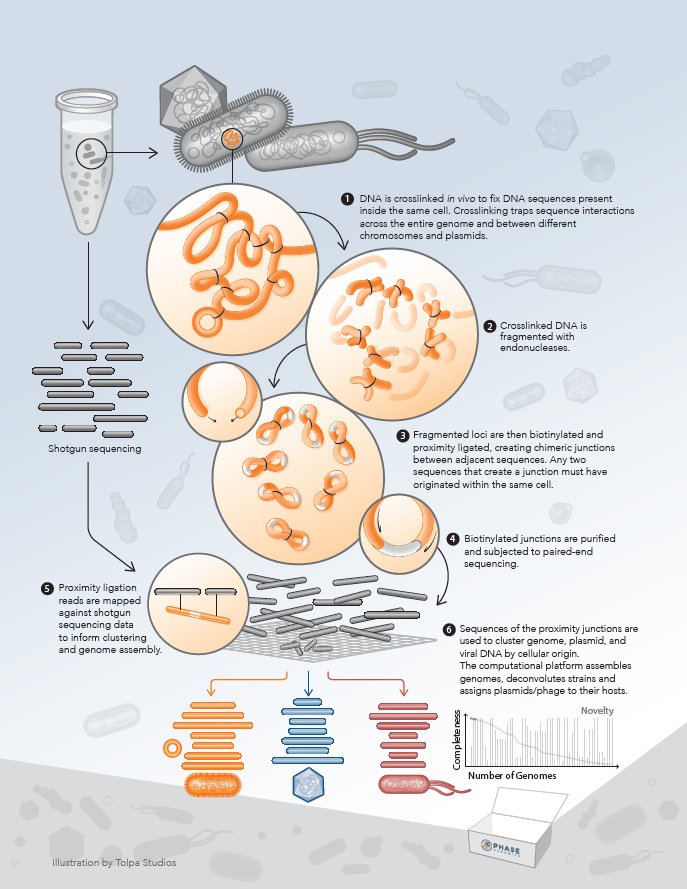
- Learn how proximity ligation enables advances in metagenomics, such as accurate viral genome reconstruction and host assignment
Proximity ligation enables ultra-long-range genome sequencing
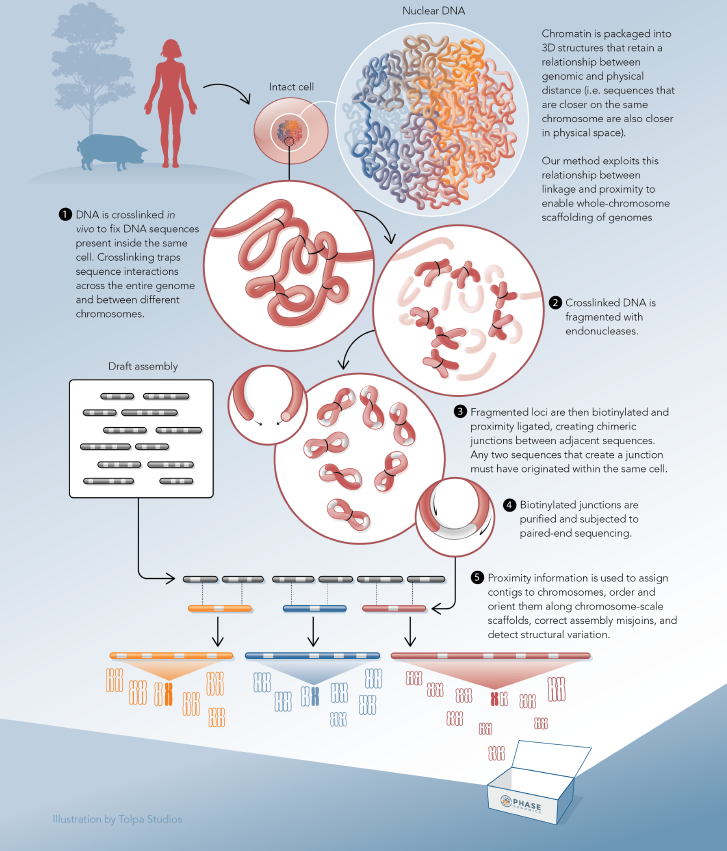
Chimeric junctions between adjacent sequences encode quantitative, long-range information
During the preparation of a proximity ligation library, DNA is crosslinked in vivo to fix the chromatin contained in the cell. Crosslinking “traps” DNA sequences that are in close proximity to one another, across the entire genome and between different chromosomes. In microbes, interactions between genomic DNA and mobile genetic elements (e.g. plasmids and transposons) are also captured. Crosslinked DNA is subsequently fragmented with endonucleases. Fragmented loci are biotinylated and proximity ligated to create chimeric junctions between adjacent sequences. Biotinylated junctions are purified and subjected to paired-end sequencing.
The information contained in these chimeric proximity junctions is not limited to sequence (position in the linear genome), but can be decoded to reveal the physical origin of each junction partner in the three-dimensional structure of the DNA. This in turn, enables improved insights in many areas of biology and medicine.