ProxiMeta Metagenome Deconvolution Platform »
Includes an 8-pack proximity ligation library prep kit, as well as on-line analysis
Viruses are ubiquitous, infecting bacteria, archaea, and eukaryotes. In microbial systems they shape the evolution and population dynamics of their hosts, both through infection and as vectors of horizontal gene transfer— thus crucially impacting the natural and human-made ecosystems in which they occur.1,2
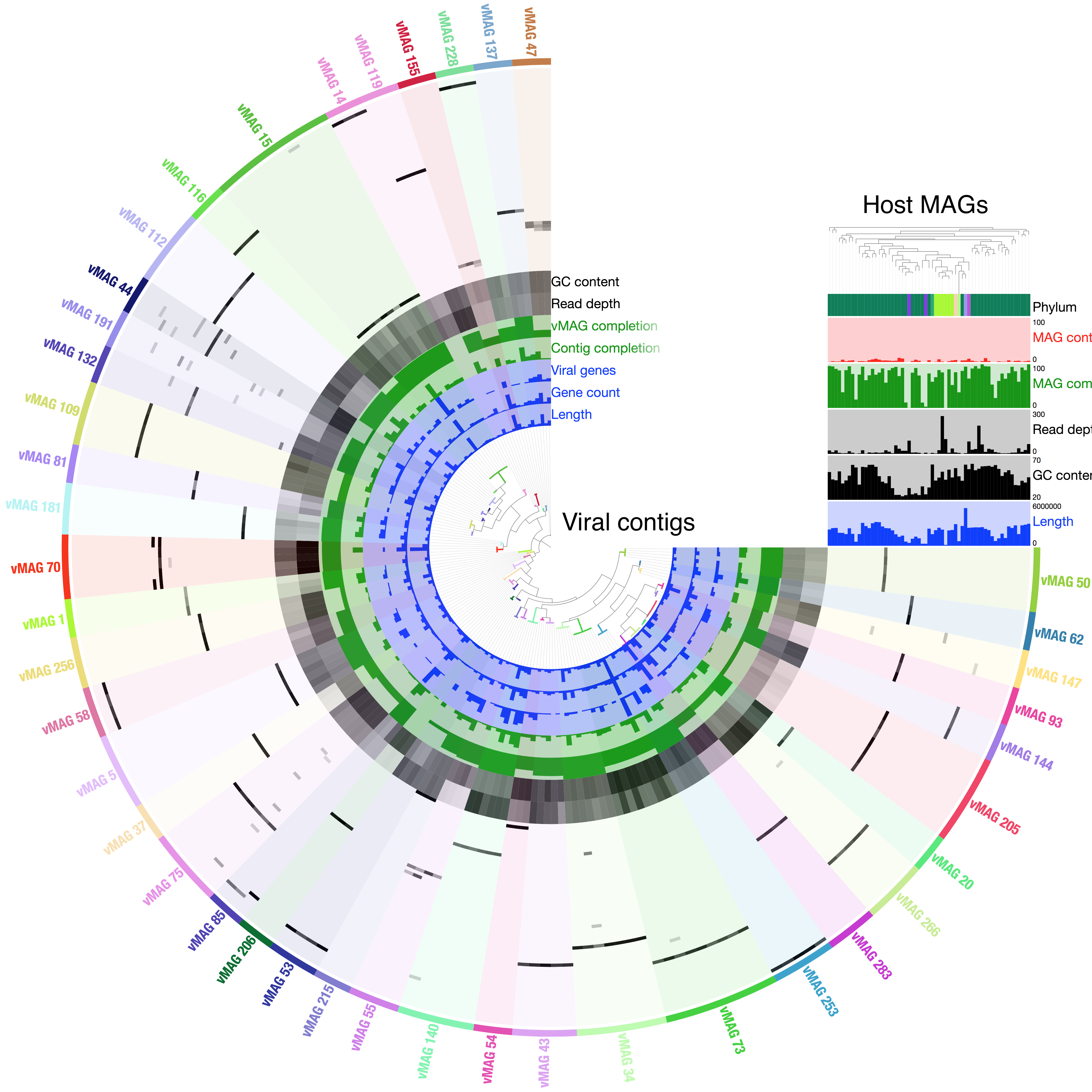
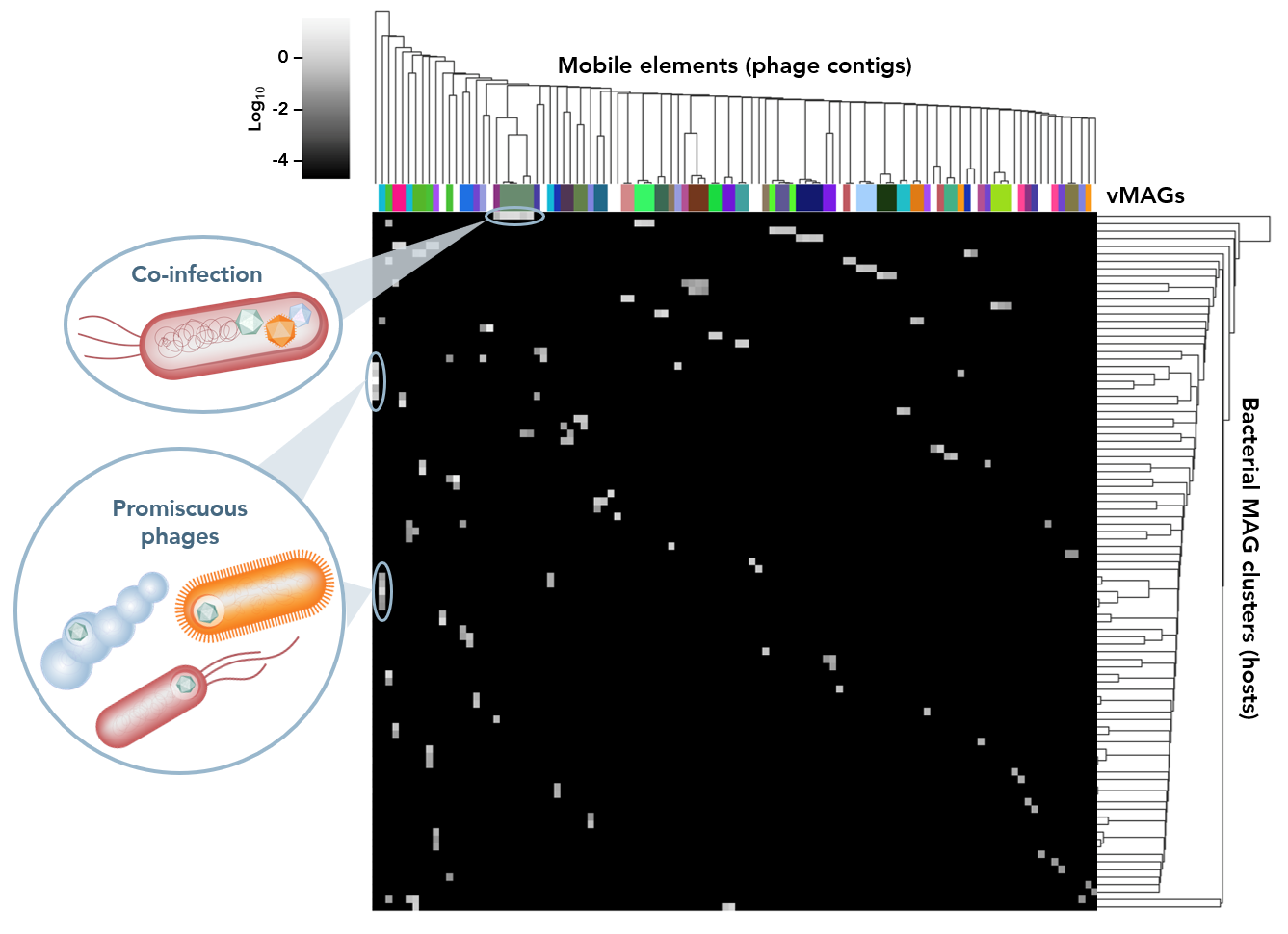
Metagenomic technologies have become key enablers in the study of environmental viromes. However, shotgun sequencing data lacks the long-range information needed to reconstruct these fragmented genomes. As a result, metagenome assembly and binning pipelines have to rely on a priori knowledge from reference databases and statistical assumptions. These methods can also not tell with certainty which sequences originated from which cell in a complex microbial community. This leaves metagenome-assembled genomes (MAGs) incomplete and contaminated. In addition, conventional binning approaches largely fail in accurately identifying bona fide hosts for the viruses in metagenomic samples, leaving major gaps in our understanding of microbial ecosystems.
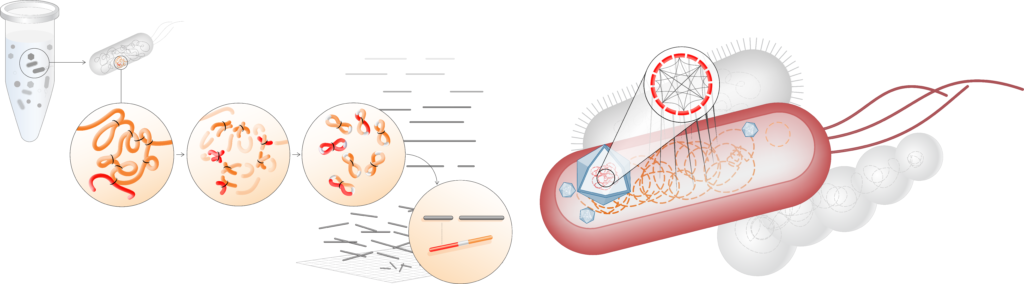
Our ProxiMeta™ Metagenome Deconvolution Platform—now powered with the new ProxiPhage™ algorithm—employs proximity ligation (Hi-C) technology to capture physical interactions between sequences within the same cell. ProxiPhage augments metagenomic analysis with this additional layer of linkage information to enable specific and sensitive host attribution of DNA viruses (phages) and reconstructs more, higher-quality bacterial and viral genomes in the process.
Download our bioRxiv preprint, Application Note and World Microbe Forum 2021 Poster to learn more about accurate viral genome reconstruction and host attribution from metagenomic samples.
ProxiMeta Metagenome Deconvolution Platform »
Includes an 8-pack proximity ligation library prep kit, as well as on-line analysis
View example reports or log in
Ultra-long-range Genome Sequencing »
Learn more about our technology