Computational Platforms
Integrate proximity ligation data to unlock an added dimension
Powerful computational tools for genomics
Phase Genomics’ proprietary cloud-based bioinformatic platforms employ novel computational approaches and algorithms to analyze and integrate proximity ligation data. This provides you with the ultra-long-range and quantitative information needed to unlock an added dimension in your cytogenomic, metagenomic, and epigenomic research.
Learn more about using our proximity ligation kits and our computational tools for applications in:
Vist our GitHub page for additional information about some of the computational tools described below.
ProxiMeta: Metagenome Deconvolution
Use the most-published Hi-C metagenome deconvolution software to make sense of your microbiome samples
Unlike whole-genome sequencing (WGS) binning methods, which rely on statistical assumptions about parameters such as k-mer frequency and coverage, Hi-C data enables metagenomic deconvolution based on true biological measurements. Unlike 16S-based analysis, Hi-C data does not rely on any a priori information to make sense of metagenomic communities, and produces proximity-assembled genomes (PAGs) for eukaryotes, prokaryotes, and archaea. Because Hi-C (proximity ligation) provides direct measurements of DNA sequences present in the same cell in vivo, the ProxiMeta Platform also enables host attribution for mobile elements, such as plasmids, phages and antibiotic resistance genes (ARGs). The platform is designed to give you more than just a taxonomic list of what is in your sample or a collection of genome FASTAs; it includes reports that provides more context, allows for genome annotation, genome comparisons, and integration with several other industry-leading deconvolution tools.
ProxiMeta Features and Benefits:
- Most published Hi-C metagenome deconvolution software
- Only commercial Hi-C metagenome deconvolution service
- Only Hi-C metagenome deconvolution software that includes comprehensive taxonomic, gene content, and novelty reporting
- Only software able to accurately associate hosts with mobile elements using raw, uncultured samples
-
- Works on eukaryotes as well as prokaryotes and archaea
- Detects cellular abundance as low as 0.02% (1 in 5,000 cells) in a variety of sample types (e.g. stool, swabs, soil, or any type of water sample)
- Able to use our most-published algorithms, or the latest open-source metagenome deconvolution software if desirable
- Custom analyses and product extensions are available
- Your data and results stored for free, forever
How do I access ProxiMeta?
- Visit proximeta.phasegenomics.com to view example reports or perform your on-line analysis (included with ProxiMeta kits).
- Contact us for assistance with your ProxiMeta analysis, or to design a metagenome deconvolution project.
ProxiMeta Analysis Workflow
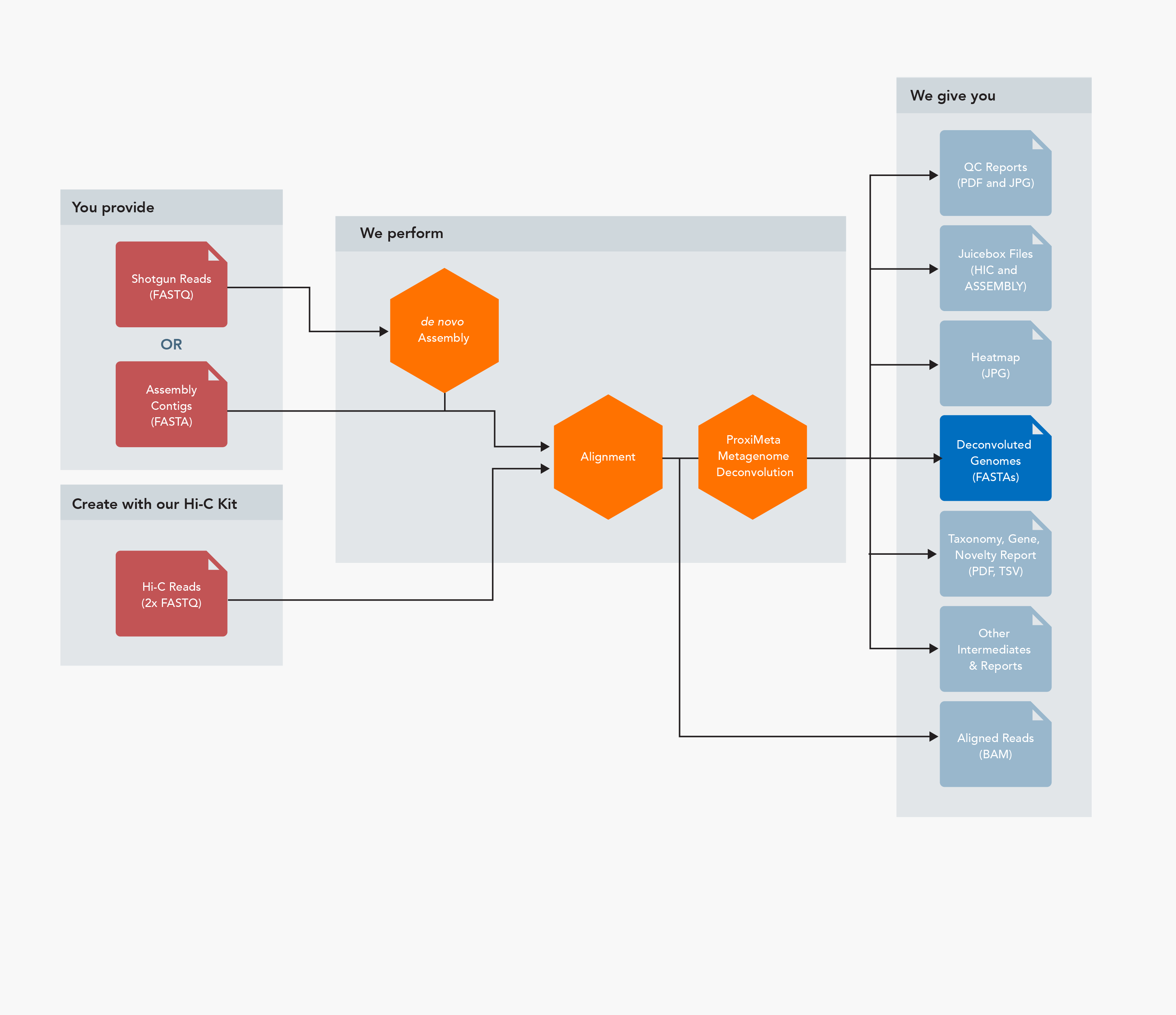 Inputs
Inputs
- Short-read shotgun read pairs (FASTQ format) or assembled contigs (FASTA format – any sequencing technology and assembler may be used)
- Hi-C read pairs (FASTQ format)
Outputs
- Individual deconvolved microbial genomes (FASTA format)
- Report detailing taxonomy, gene content, novelty relative to RefSeq, and other statistics (PDF and TSV format)
- Juicebox files (HIC and ASSEMBLY formats)
- Aligned Hi-C reads (BAM format)
- QC reports (PDF and JPG formats)
- Various intermediate files and reports
Proximo: Genome Scaffolding
Three-stage scaffolding algorithm and scaffold optimization process for the best, most complete scaffolds
Proximo assigns contigs to scaffolds, arranges contigs into a linear ordering, and then orients contigs in such a way as to maximize the likelihood of having generated the observed Hi-C data. This core scaffolding algorithm is combined with a scaffold optimization process that performs tens or even hundreds of thousands of scaffolding attempts in order to find the scaffold solution most concordant with the data. Proximo is also the only Hi-C scaffolding algorithm capable of directly consuming linkage maps or reference genomes, providing the ability to use more of your data as input to generate the best possible scaffolds.
Proximo Features and Benefits:
- Most-published Hi-C scaffolding tool
- Typical scaffolds include ≥98% sequence length on chromosomal scaffolds; very rarely less than 95%
- Able to identify and break misjoined contigs
- Scaffolds will have all polishing steps performed – open source tools leave a lot of manual work remaining
- Guaranteed correct number of scaffolds – open source tools often still leave genomes in thousands of pieces
- Able to use linkage maps and/or reference genomes as inputs (if available and desired)
How do I access Proximo?
Contact us to design or get started with a Proximo Genome Scaffolding project.
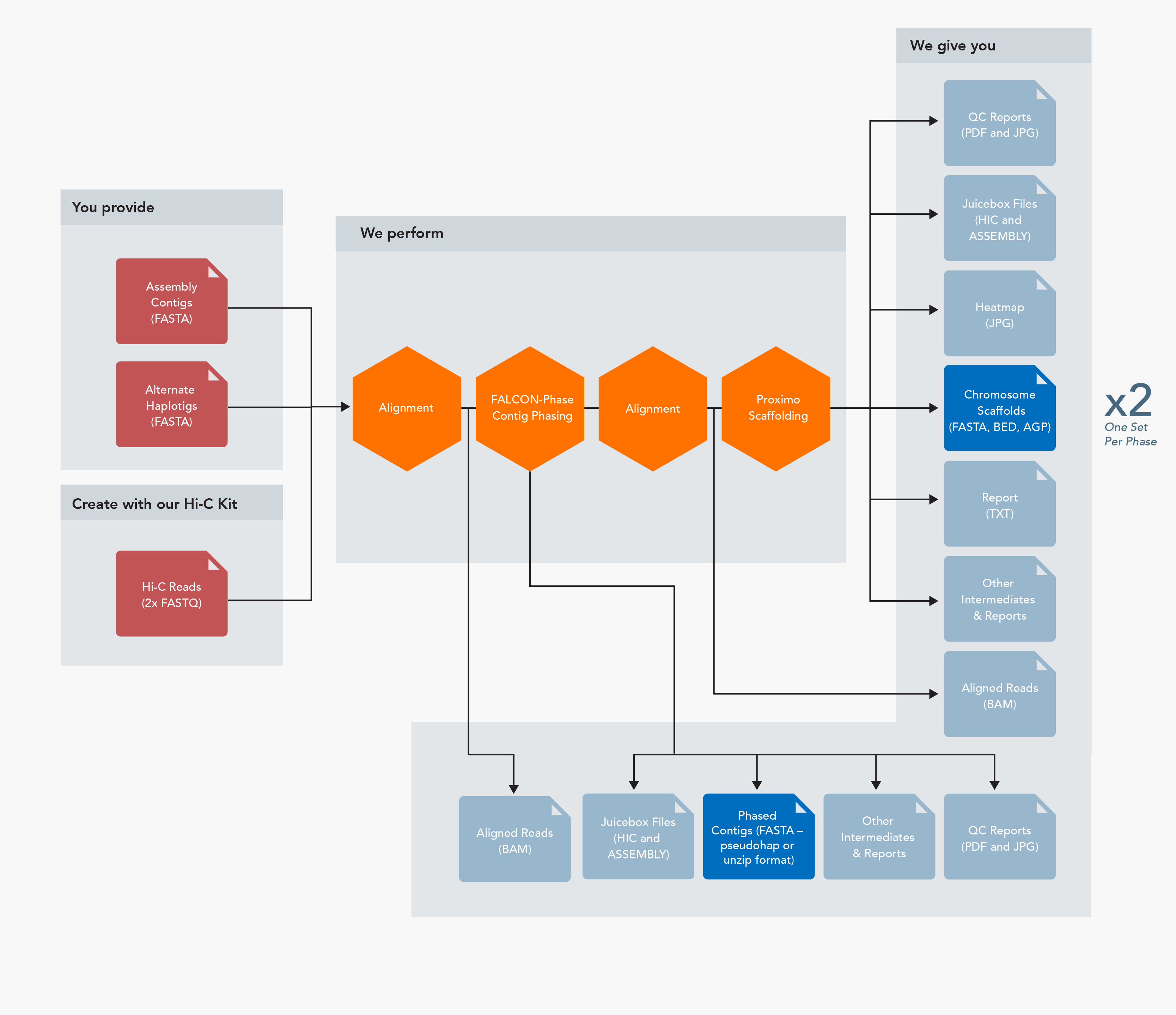
Proximo Analysis Workflow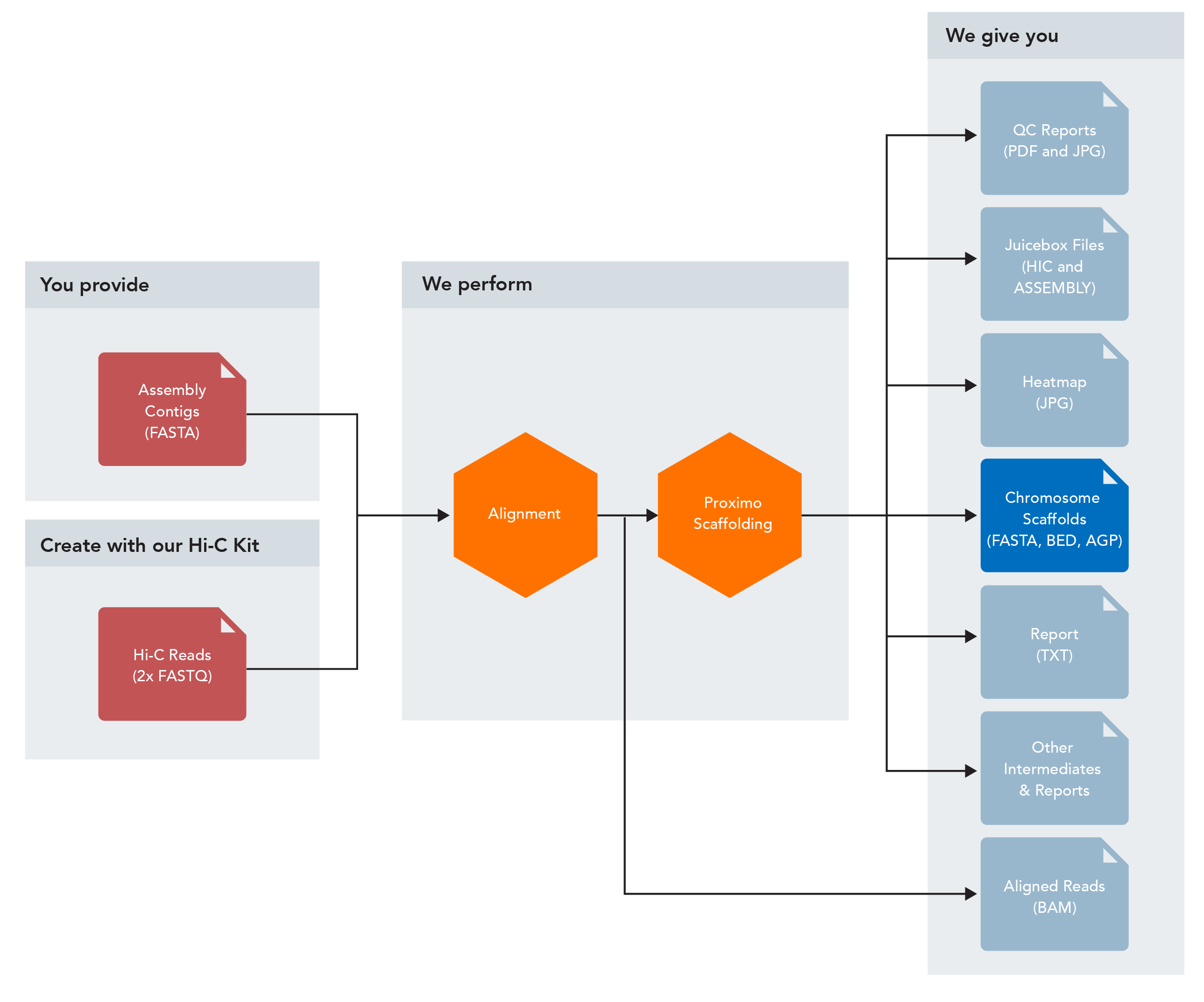 Inputs
Inputs
- Assembled contigs/intermediate scaffolds (FASTA format)
- Hi-C read pairs (FASTQ format)
- Linkage map (optional – 2 column format or equivalent)
- Reference genome (optional – FASTA format)
Outputs
- Chromosome-scale scaffolds (FASTA, BED, and AGP formats)
- Juicebox files (HIC and ASSEMBLY formats)
- Aligned Hi-C reads (BAM format)
- Scaffold heatmap (JPG format)
- Summary report (TXT format)
- QC reports (PDF and JPG formats)
- Various intermediate files and reports
FALCON-Phase: Haplotype Phasing
Obtain complete, fully phased diploid scaffolds
Combine Hi-C data with long reads to create two true phased sets of contigs for diploid organisms. Use FALCON-Phase in combination with Proximo to produce complete, fully phased, diploid scaffolds.
Sister chromatids are independent DNA molecules in the nucleus, and as such they form independent Hi-C profiles that can be used to identify which heterozygous sequences originated on the same molecule. FALCON-Phase examines contigs and haplotigs (e.g., the results of FALCON-Unzip or purge_haplotigs) in the context of Hi-C data, using a graph partitioning algorithm that detects likely phase switch errors and corrects them. This results in >96% contig phasing accuracy in known-truth, pedigree-based benchmarks. FALCON-Phase can also be used in conjunction with Proximo to extend phase blocks to the chromosome-scale, delivering two complete, true-phased sets of chromosomes for diploid organisms: the paternal genome and the maternal genome both, from a single analysis.
FALCON-Phase Features and Benefits:
- Only Hi-C based phasing software available
- Able to integrate with Proximo and produce complete, fully phased diploid scaffolds
- Compatible with PacBio’s FALCON assembly stack, or with any other long-read technology provided that purge_haplotigs is run
Unlock the diploid human genome
Learn about FALCON-Phase, and how it was used to combine PacBio long-read and Hi-C data to produce the most contiguous diploid human genome assembly.
How do I access FALCON-Phase?
- FALCON-Phase is open-source. Visit github.com/phasegenomics.
- Contact us to design or get started with a FALCON-Phase project
FALCON-Phase Analysis Workflow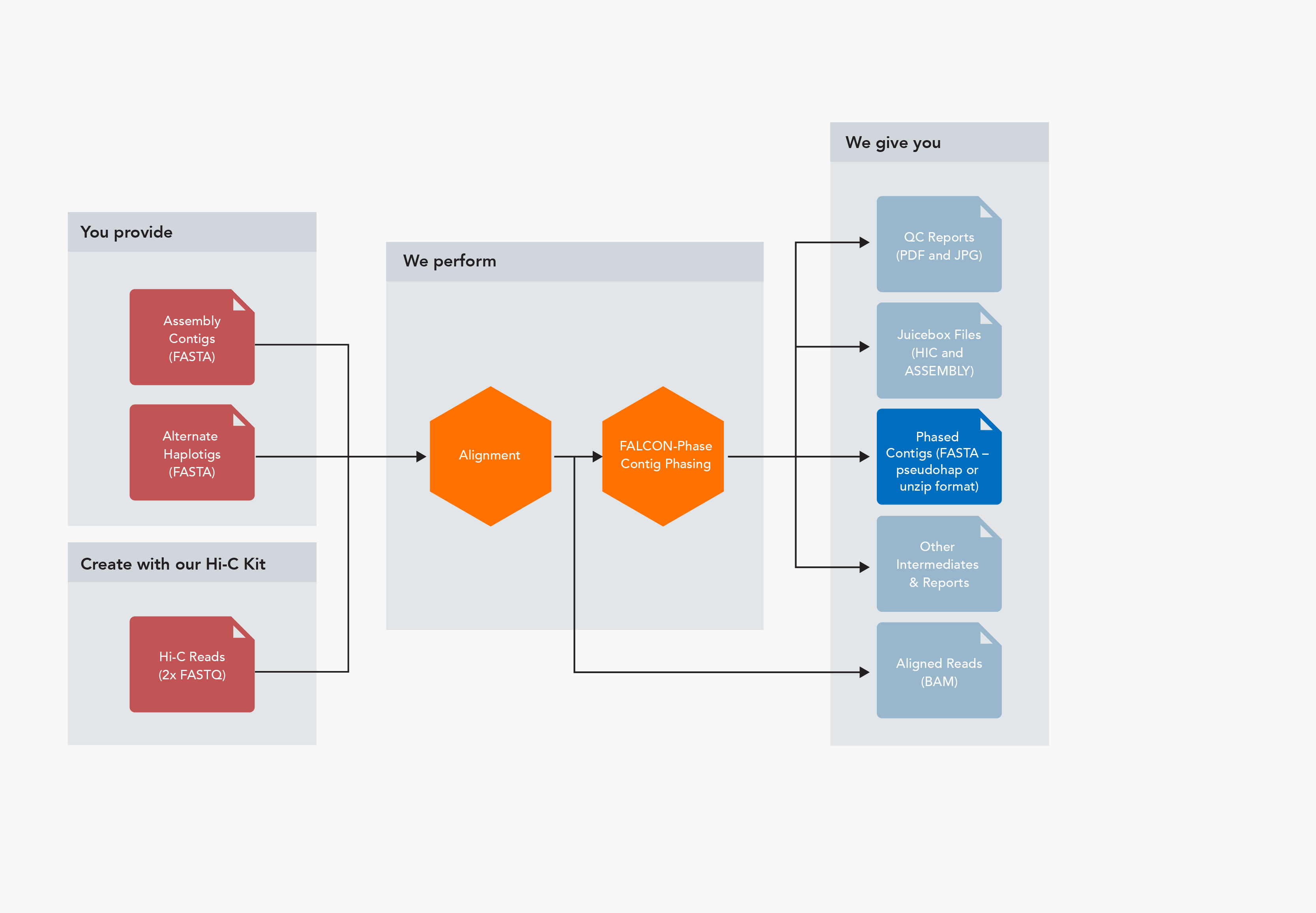 Inputs
Inputs
- Primary contigs (p_ctgs) and alternate haplotigs (h_ctgs) (FASTA format)
- Both FALCON-Unzip and purge_haplotigs produce the proper files
- Hi-C read pairs (FASTQ format)
Outputs
- Phased contigs – two formats available
- Pseudohap – each phase is a complete set of contigs (FASTA format)
- Unzip – primary contigs and alternate haplotigs, phase switch errors corrected (FASTA format)
- Juicebox files (HIC and ASSEMBLY formats)
- Aligned Hi-C reads (BAM format)
- QC reports (PDF and JPG formats)
- Various intermediate files and reports
- When combined with Proximo, one set of all Proximo outputs for each phase (see below)