
Phase Genomics partnered with Browne Family Vineyards to begin to understand, the microbiome makeup of soils within different vineyards across the state of Washington. The findings were unveiled at the Pacific Science Center’s “STEM: Science Uncorked” winetasting event.
There are many different factors that contribute to soil composition, such as parent material, topography, climate, geological time, and the thousands of different and undiscovered microbes living in the soil—the least understood factor. In April of 2018, Browne Family Vineyards staff visited five of their vineyards, filled a bag with soil from each site, and sent it to Phase Genomics to analyze the microbiome in each of the soil samples.
SYMBIOSIS BETWEEN PLANTS AND MICROBES
Plants rely heavily on their microbiome to live, grow, and protect themselves from pathogens. One example of this symbiotic relationship is that plants release chemicals into the soil in order to attract microbes. These microbes bring nutrients such as nitrogen, iron, potassium, and phosphorus to the plants in exchange for sugar, which the microbes require to survive. Microbes also play an important role in nitrogen fixation, organic decay, and biofilm production to protect the plant roots from drought. It is evident that this symbiotic relationship between microbes and plants is critical to the health and survival of both, but further research into this complex community is inhibited by two main problems: It is impossible to isolate microbes in such a complex mix and most of the microbes have never been discovered before.
THE DARK MATTER OF THE MICROBIOME
Microbes live in communities where they rely on each other. This makes it difficult to isolate or culture (i.e. grow) microbes without killing them or altering their genetic makeup. Moreover, there can be millions of microbes living in a single teaspoon of soil, making these samples extremely complex environments. This causes most of the microbial world to be unknown, sometimes referred to as the “Dark Matter of the Microbiome”.
The most effective way to identify the microbes in the community is to look at the genetic makeup of the microbiome to try to classify microbial genomes present. Standard practices include sequencing of 16S (a hypervariable genomic region) and shotgun sequencing. By combining these standard practices with Hi-C, researchers are now able to fully reconstruct genomes from a mix because Hi-C captures the DNA within each microbe to exploit key genetic features unique to each individual in the community. The Phase Genomics Hi-C kit and software, ProxiMetaTM, uses this information to capture even novel genomes straight from the sample without culturing—illuminating the dark matter of the microbiome.
THE PROCEDURE
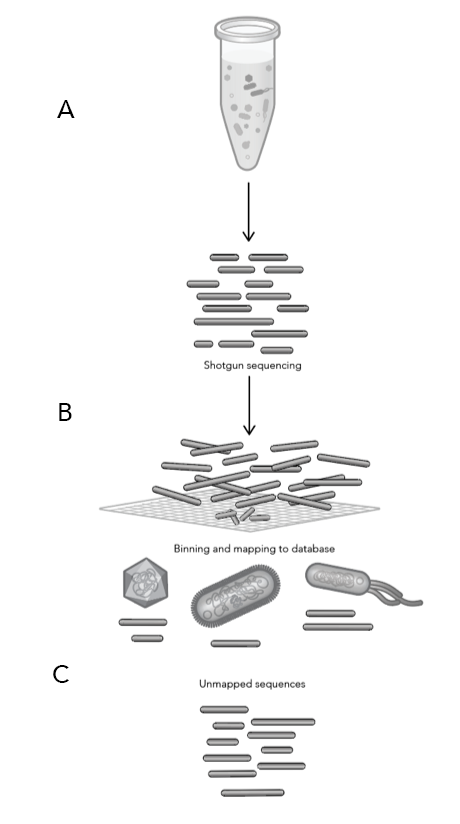
Figure 1: Shotgun Sequencing Procedure and Difficulties
Once the soil samples were collected from the five vineyards, Phase Genomics produced shotgun libraries to obtain DNA from all of the microbes in each sample (Figure 1)—essentially taking the soil sample, breaking open all of the microbial cells then purifying the DNA (1.A). Since DNA is fragile, most of it gets broken into smaller pieces during this process, leaving a mix of many DNA fragments from all of the microbes that were present in the original soil sample. The fragmented DNA is then sequenced and the “sequence reads” are uploaded into a database of known microbial genomes (1.B). This database then searches for matches or “hits” to see if the reads are similar to anything in the database (1.C).
A problem with relying on shotgun data is that it’s unclear which DNA fragments belong to which microbe, thus relying heavily on computational techniques and the accuracy of the reference database for classification. This results in little improvement or clarity on the makeup of the sample, again, leaving the microbiome in the dark. Though shotgun sequencing only provides a glimpse into the microbial community, this data allows scientists to differentiate the taxonomy (phyla, genera, species) of the microorganisms living in the soil.
THE RESULTS
Shotgun sequencing identified over 10,000 different species from each of the vineyard soil samples; however, it is impossible to know if this is the true number of species because only ~ 20% of the reads matched the database, indicating ~80% was either incomplete or undiscovered (see table below).
| Vineyard | Total Reads | Percent of Reads Classified | Number of Organisms Found | Percent of Unknown Organisms |
| Canyon | 19,001,222 | 15.95% | 10,726 | 73.32% |
| Canoe Ridge | 21,214,190 | 17.66% | 11,721 | 55.55% |
| Waterbrook | 19,469,954 | 19.6% | 10,782 | 50.58% |
| Skyfall | 63,850,810 | 16.17% | 15,101 | 80.08% |
| Willow Crest | 43,941,026 | 17.13% | 13,914 | 71.84% |
Moreover, of assigned reads, >50% did not match to a genus or species—hinting that many of the organisms found are novel. Without digging too deep into the microbiome analysis, it is evident that the microbial makeup is different for each of the samples. Varying levels of reads from each vineyard were able to be classified (Table 1), and among the classified reads, the vineyards have 3-4 microbes that vary in abundance in common. These microbes, such as Proteobacteria, Rhizobacteria, and Actinobacteria, generally, are very common in soil.

Proteobacteria
There are obvious differences in the biodiversity of the soil samples both in number of species and relative abundance. For example, Canoe Ridge and Waterbrook samples were >20%, Delftia, while the microbes in the other vineyards were more evenly distributed, with abundance closer to 1-5%. Interestingly, Delftia, a rod-shaped bacterium, has the ability to break down toxic chemicals and to produce gold.

Actinobacteria
There are two main components that influence microbe classification in these samples: the desired taxonomy level, and the statistical threshold, or minimum number of reads, set to define it. Much like zooming in and out, the most “zoomed out” analysis is achieved by a stringent threshold and will reveal phylum, while the most “zoomed in” analysis is achieved by a more lenient threshold and will reveal genus and species
If the data is “zoomed in” further, about 37% of the microbes in each community can be identified by genus. On average, 63% of the communities do not match to a genus at all, hinting that these microbes may have never been sequenced. The most abundant microbe genera present in these samples are Bradyrhizobium, Streptomyces, and Nocardiodes.
As discussed earlier, this data highlights the issues that are present with shotgun data and the corresponding analysis: there is still far too much that is unknown. In order to better understand these samples, we also performed Hi-C on two of the samples which will be discussed in further detail in the next section.



HI-C AND FINDING NOVEL GENOMES
One thing all these soil samples have in common is that they are composed of numerous novel species. To obtain more information on the microbes present in these samples, and solve the issue discussed earlier surrounding shotgun data, Hi-C was performed on two of the soil samples, Skyfall and Willow Crest. Essentially, Hi-C assigns DNA fragments from shotgun sequencing to the correct species by connecting DNA while the cells are still intact.
Hi-C enables clustering of shotgun assemblies and subsequently yields complete genomes from a microbiome, even if the genome has never been sequenced before. With complete microbe genomes, it becomes easier to classify organisms down to the strain-level—a step even further than species. By having the genome, we can essentially read a microorganism’s blueprint and learn more about its genes, evolution, and even function once the genome is annotated.
For example, preliminary data from the Willow Crest soil sample yielded 400 different genome clusters. When compared to known bacterial genomes in the RefSeq database, which aggregates all published microbial genomic data, over half of the extracted genomes are unable to be identified at a genus level and thus likely represent newly discovered bacterial organisms.
SCIENCE UNCORKED
When the microbiome data from the vineyards were presented to the public at the Pacific Science Center, two questions consistently arose: How does this influence wine taste, and how can growers select for a healthy microbiome? These very forward-thinking questions unfortunately cannot be answered—yet.
Scientists do know that soil plays a big role in plant health, and this could in part be due to the plants’ symbiotic relationship with microbes, as discussed earlier. It has also been shown that biodiversity can benefit plants because of the diverse functions individual microbes have, i.e. with more microbes, there are more potential functions being served versus 1 microbe serving one function. However, nailing down answers to these questions will take a lot of research. With emerging technologies, like Hi-C, the answers have become much more obtainable.
Though the term “microbiome” may not be household vocabulary, many of the attendees were very aware about the role that microbes play in human health, and how they influence the world around us. It goes to show that the rapid developments in the microbiome field are reaching beyond just research and becoming more tangible for the general public. Relevant stories—like looking into the microbiome of vineyards— are helping them understand the intricate concept of microbial life.
Learn more about ProxiMeta Hi-C and the microbiome by visiting our website www.phasegenomics.com and connect with us on twitter by following @PhaseGenomics