Genome sequencing has confirmed some long-held theories about the blueprints of life. But it has also unearthed quite a few surprises. Scientists once hypothesized that the human genome consisted of upward of 100,000 genes. The decades-long Human Genome Project — as well as many next-generation sequencing studies — have prompted the downward revision of that figure to a relatively spartan 20,000 genes, more or less.
Evolution in action
If there is a lesson in this vast overestimation to our gene load, it is perhaps that evolution shapes genomes in unexpected ways.
The advent of more nimble and lithe methods for genome assembly and analysis holds the promise to unearth the surprises that evolution has wrought. These relatively new advancements include tools like Phase Genomics’ ultra-long-range sequencing, which reconstructs the sequence of chromosomes by using positional relationships between DNA sequences in the genome. These methods have grown sufficiently sophisticated to catch the quick transitions that transform populations and species.
Recently a team led by Dr. Leonid Kruglyak at UCLA employed these tools to catch evolution at work. Their discovery relates to sex determination, a complex developmental process that, in animals, generally kicks off when an immature gonad develops into either testes or ovaries. In humans and many animals, sex determination is governed largely by genes, and in turn shapes their genomes and evolutionary trajectories like few other biological processes can.
That special pair
For species with full genetic control over sex determination, the process often leaves its imprint on the genome in the form of sex chromosomes. In most animals, genomes consist of pairs of chromosomes called autosomes. But in addition to those autosomes, many animals — including us — harbor another set of chromosomes called the sex chromosomes. Sex chromosomes govern — or at least try to govern — whether the gonads develop into ovaries or testes, which in turn influences the development of genitals and secondary sex characteristics.
Scientists have long theorized that sex chromosomes evolve from autosomes. Studies of young, relatively new sex chromosome systems, like those in the medaka, indicate that the transition happens fast. Yet the steps that transform a pair of autosomes into sex chromosomes are at best murky, with many questions unresolved. Much could be answered by catching this transition from autosome to sex chromosome in the act.
Behind the curtain
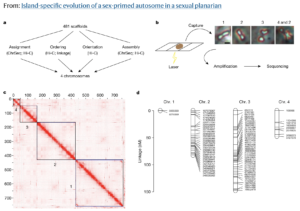
In a paper published June 1 in Nature, Dr. Kruglyak and his colleagues announced that they have found just such a transition: an animal with a pair of autosomes that is beginning to act like sex chromosomes. The researchers utilized Phase Genomics’ Proximo™ genome scaffolding platform and PacBio long reads to sequence and assemble a highly complete genome for a microscopic, freshwater flatworm, Schmidtea mediterranea. In many parts of its natural habitat across the Mediterranean basin, S. mediterranea reproduces by budding, without the need for sex. But some populations in Corsica and Sardinia produce the next generation through sexual reproduction.
The team, including lead and co-corresponding author Dr. Longhua Guo at UCLA, discovered that in these sexual strains of S. mediterranea, one pair of autosomes shows evidence of almost no genetic exchange, also known as recombination, during reproduction. This is a telltale signature of sex chromosomes. In addition, they saw that the unusual pair of autosomes harbors a large contingent of genes that play a role in developing sex-specific characteristics. Taken together, these genomic data finger these autosomes as a “sex-primed” pair that are in the process of evolving into fully fledged sex chromosomes.
Photo finishes
Future studies of S. mediterranea’s nascent sex chromosomes will likely fuel fresh inquiry and debate about this rarely-seen evolutionary transition. The answers will stretch far beyond flatworms. Studies of other recently evolved systems, such as in stickleback fish, show that sex chromosomes can play a decisive role in other poorly understood evolutionary transitions, such as the rise of a new species.
Beyond sex chromosomes, this study demonstrates the raw interrogative power of modern genome assembly and analysis methods. They can capture transitions — even the most brief and ephemeral. Applied appropriately, methods like these can help scientists make sense of a myriad of messy, complex processes that evolution shapes. These include some issues that hit as close to home as gonads, from curbing the spread of antibiotic resistance to protecting pollinators from annihilation. Evolution moves quickly. Now, so can we.






 LinkedIn
LinkedIn Email
Email