
This month Phase Genomics is celebrating #FungusFebruary by highlighting some of the unique capabilities of our Hi-C technology to solve age-old mysteries in the world of fungal genetics and deliver new potential for researchers to understand fungi, all while helping solve global crop crises and develop new groundbreaking pharmaceuticals.
While we wield the power of genomics to explore the wonders of fungi today, a few centuries ago people dismissed them as just weird plants. Eventually microscopes and anatomical studies revealed fungi as a distinct flavor of life — some varieties quite tasty — but educational experts today continue to bemoan the lack of lessons on fungi in biology curricula, and research on fungi — even those that cause disease — lags.
As a result, scientists lack much basic information on the genetics, life cycles, and reproductive habits of many fungi — even though members of this kingdom could help address a bevy of challenges in food and energy production, illuminate the evolution of complex life and even shelter us on Mars.
Genome studies on fungi of all stripes can resolve evolutionary relationships and ecosystem dynamics, identify metabolites of commercial and medical interest and — for fungi that cause disease — reveal biochemical and genetic targets to help us fight pathogenicity.
Like their animal and plant cousins, fungal genomes also have their challenging parts, including repeats, duplications and structural elements that complicate both sequencing and assembly. Recently, the chromosome conformation method “Hi-C” and advances in next-generation sequencing have helped untangle some of these sticky genomic knots, and show promise in taming genomes across this diverse and neglected kingdom of life.
1. High-resolution mapping of centromeres
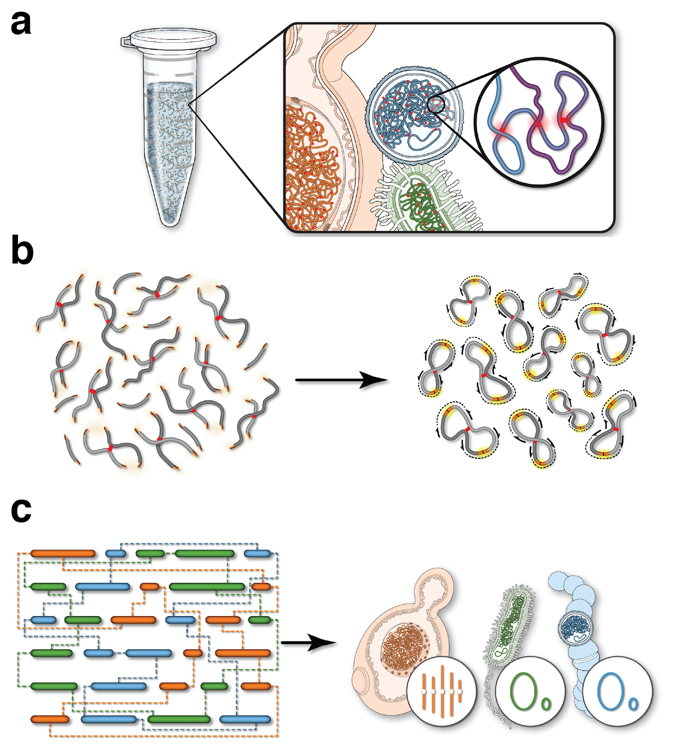
Hi-C’s power lies in its ability to identify regions of the genome that reside in close proximity to one another in the nucleus — information that essentially captures the 3D organization of the genome. But Hi-C doesn’t just identify where particular chromosomes reside within the nucleus. It can also help identify functional elements in genomes that are difficult to identify in other ways.
That is what two groups of researchers (from the Pasteur Institute and the University of Washington) did when they used Hi-C to track down functional elements in yeast genomes — centromeres and rDNA clusters — both of which are typically repeat-rich and difficult to identify without laborious experiments involving functional assays or mapping the binding sites of rare centromere proteins. In fungal species, centromeres are held tightly together at the spindle pole body, and the team used this shared proximity to identify centromere locations in the genomes of numerous yeasts (and subsequently other fungi), despite not knowing their centromeric DNA sequence. Ribosomal DNA clusters similarly congregate in yeast nuclei, which one team exploited to identify their positions in Debaryomyces hansenii.
2. High-quality genomes illuminate biochemical pathways
Fungi harbor a wide array of genes for synthesizing secondary metabolites, which range from harmful toxins to helpful pharmaceuticals. In fungi, genes for synthesizing secondary metabolites tend to occur in clusters, which are also thought to be sites of rapid evolution.
Phase Genomics worked with a University of Minnesota-led team and used Hi-C to generate high-quality genomes of six strains of Tolypocladium inflatum, an insect pathogen that has already given us the immunosuppressant drug cyclosporin. The new assemblies revealed major differences in secondary metabolite production between T. inflatum strains, including novel clusters, transpositions and clusters that may be involved in toxin synthesis. The bevy of discoveries from these assemblies showed how recombination can drive significant divergence even within a single species — and how important it is to build multiple high-quality genome assemblies that can capture that diversity.
3. Fungal dikaryons and the hidden nuclear dance
The genetic differences between strains also apply to pathogenic fungi, like the stem rust, which parasitizes wheat. Phase Genomics partnered with a team led by scientists at CSIRO in Australia to apply Hi-C to stem rust – the particularly deadly scourge Ug99. Like many fungi, stem rust genomes are divided between two haploid nuclei. The team used Hi-C data to assemble complete haplotypes for both haploid genomes of both strains, and discovered that Ug99, a recent arrival that is decimating whole fields of wheat in Africa, has an unexpected origin: The strain arose through “somatic hybridization,” when hyphae from two strains exchange haploid nuclei. This may explain the strain’s sudden rise and deadly wake, and gives scientists new genomic information to understand Ug99’s virulence and identify weaknesses that could give wheat a leg up.
4. Hybrids, beer, and fungal metagenomics
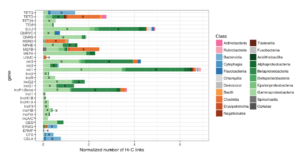
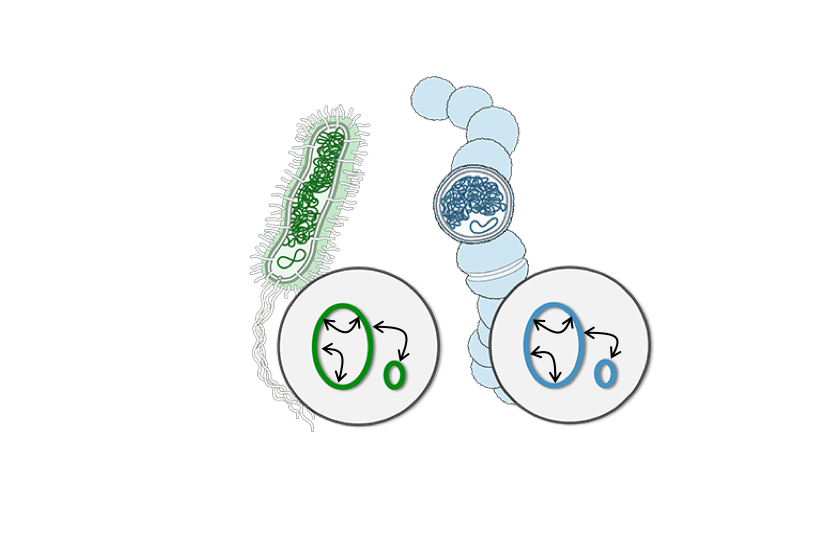
The ability to separate two nuclei from within the same cell can be extended to more complex samples. Yeasts, which are integral players in brewing, will often hybridize to form new species containing genomes from two organisms at once (the famous lager-producing yeast Sacharomyces carlsbergensis is one example of such a hybrid). But in a mixed microbial community, such as beer, wine, or a microbiome sample, how can DNA sequencing detect which genomes co-exist within the same cell? One special power of Hi-C is that it traps sequences that are within touching distance of each other, and therefore must come from inside the same cell. The Dunham lab at the University of Washington used this property to analyze an open-fermentation beer from a local brewery. The exciting result was that they were able to discover a new hybrid yeast, later named Pichia apotheca, using Hi-C data to identify it as a hybrid bearing two genomes from related organisms. This new hybrid species has since been used by home-brewers to ply their craft and gives beer a very unique flavor.
5. The Epigenetics of Symbiosis
Nature has plenty of examples of plants and fungi getting along. One of them is Epichloë festucae, a filamentous fungus that has evolved a symbiotic relationship with certain grass species. When Phase Genomics worked with a Massey University-led team, they discovered that E. festucae’s genome carries hallmarks of this symbiosis. The analysis of Hi-C data revealed that important genes are clustered into blocks separated by repeat-rich regions. Hi-C and RNA-seq data together showed that genes within the blocks have similar expression patterns — indicating that genes needed for symbiosis with their grass hosts tend to cluster together in the same blocks.
Looking Forward
Cutting edge genomic technologies like Hi-C have the potential to keep making up for lost time and reveal even more intimate details of the hidden lives of fungi. This #FungusFebruary, it’s worth asking: What other mysteries about this long-overlooked kingdom are worth solving?
































 LinkedIn
LinkedIn Email
Email