
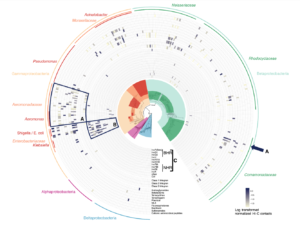
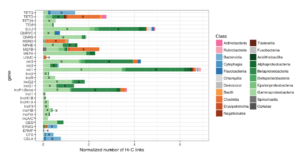
Phase Genomics continues to pioneer genomic innovation, driving advancements in human health and life science research over another impactful year. Our efforts range from developing novel tools for detecting chromosomal abnormalities to managing the world’s most extensive phage-host interaction atlas, thereby accelerating genomic research and promoting a healthier future.
Our ultra-long-range sequencing technology is fostering advancements across a wide array of research applications, both at Phase Genomics and in laboratories worldwide. Utilizing our microbial platform, we are driving transformative discovery in metagenomics and ecology while making bounds in human health with cutting-edge approaches to antimicrobial research and oncology.
Thank you to our supporters, collaborators, and clients for their contributions to making this an outstanding year. Here are some key highlights from 2024.
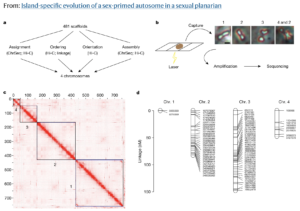
Insights in oncology

Cow burps, super bugs, and our enemy’s enemy–phages

A novel approach to vaccination

Diving into the data
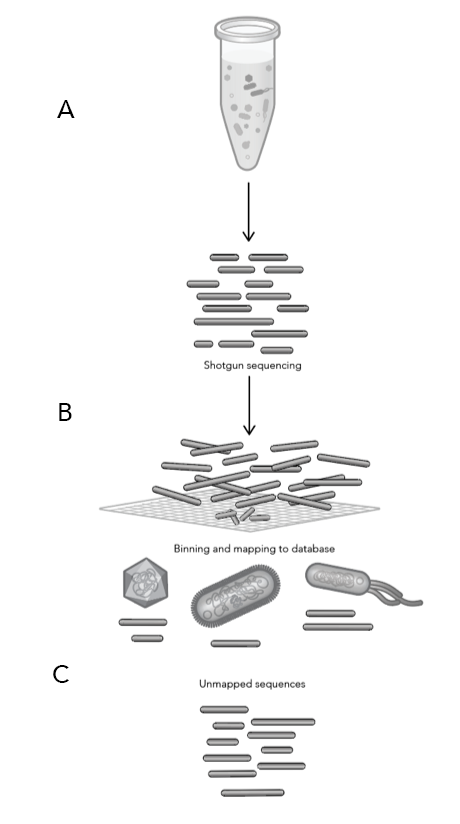
Two new data analysis tools were made available to ProxiMeta and CytoTerra platform users this year: ProxiMeta™ Explorer and CytoTerra® Curator.
ProxiMeta™ Explorer is an interactive, cloud-based genome-resolved metagenomic analysis platform for data visualization and exploration. The platform provides fully customizable analyses and reports for tracking genomes across time, conditions, groups, and more with a click of a button.
CytoTerra® Curator enables users to effortlessly review, revise, and generate reports from cytogenetic data – no prior bioinformatics experience required. From curating calls to constructing circos plots, Curator provides fast, accurate insights for human genomics and oncology research.
Tune in to this year’s podcasts
Listen to Phase Genomics CEO, Ivan Liachko, discuss the breadth of applications supported by Phase Genomics’ ultra-long-range sequencing technology in these podcast episodes. Discover the story behind commercializing and implementing biotech innovations and get a glimpse at where this technology is taking us.
- Biotech Insights – Biotech CEO Podcast
- Lab Rats to Unicorns – From Genes to Genomes: The Courage to Innovate with Dr. Ivan Liachko
Looking Forward
In 2025, we aim to elevate our technology to new heights and broaden our impact across science and medicine. We hope you will follow us on our journey on X, LinkedIn, and BlueSky as we lead genomics innovation to an insightful and healthy future.
Happy New Year from our team at Phase Genomics!





























 LinkedIn
LinkedIn Email
Email