
New genomic technology reveals the parental past of “ancient asexuals,” paving a route to crop engineering and soil remediation with symbiotic fungi
In a warming, crowded world, we need more help than ever from plants. But maximizing the bounty from crops — from food to fuel to fibers — means coaxing plants to draw minerals and nutrients from soil more effectively, and paying special heed to the tiny, often-overlooked fungi that make this possible.
Plant roots have symbiotic relationships with fungi that stretch back eons. For example, arbuscular mycorrhizal fungi, or AMF, have been cozying up to plant roots for at least 400 million years. In exchange for carbon-rich lipids from their hosts, AMF — named for the branch-like structures their bodies form within plant roots — help host defenses against pathogens, deliver water and increase absorption of nutrients rich in nitrogen, potassium and phosphorus. They also boost plant diversity.
Thanks to this ecological generosity, AMF are used as crop stimulants and in soil remediation. Their lipid lust also makes them good at carbon sequestration. Theoretically, engineered AMF strains could mount an even better performance in these essential tasks. But scientists have long viewed certain features of AMF, particularly their genetic structure and life cycle, as evolutionary puzzles that must be solved to make strain engineering possible and build better symbionts.
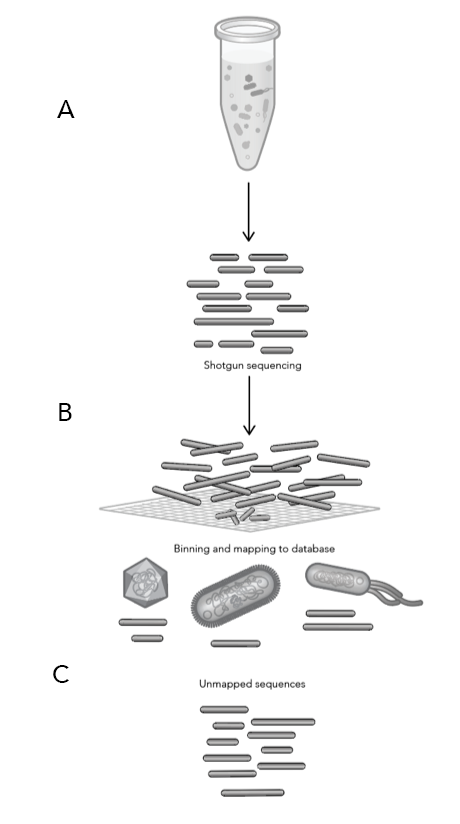
Working with Phase Genomics, scientists at the University of Ottawa recently overcame this barrier, successfully sequencing the genomes of four strains of the most widely studied AMF species, Rhizophagus irregularis. Using Phase’s proximity-ligation sequencing techology, they showed for the first time that the genomes of AMF are simultaneously more straightforward and more surprising than many mycologists had dared to dream.
Armed with this knowledge, scientists can plan new approaches to engineer AMF strains for applications in biomass production, soil remediation — and beyond.
The mysterious kary carryall
For years, the more scientists looked at AMF, the more questions they had. AMF bodies are essentially bags of haploid nuclei — tens of thousands, all sharing a common cytoplasm. And that’s not all.
“There were many, many outstanding questions about AMF,” said Dr. Nicolas Corradi, leader of the University of Ottawa team. “This was primarily because these fungi are always multinucleated and lack observable sex. It was suggested that AMF have an ‘oddball’ genetics and evolution.”
They were assumed to be “ancient asexuals,” who must’ve somehow thrived without the gene-shuffling benefits of sexual reproduction.
Dr. Corradi and his colleagues were determined to find out if that’s the case, and in the process began to shatter AMF’s asexual reputation. In 2016, they showed that Rhizophagus irregularis strains harbor evidence of sexual reproduction, including finding some of the genes needed for it. In some strains, all nuclei were genetically identical. But other, more robust and resilient AMF strains — termed heterokaryons — harbored two distinct populations of nuclei in their cytoplasm. More recently, Dr. Corradi and his team reported that the two populations of nuclei in heterokaryons change in abundance, depending on their host plant.
“But these were, however, based on fragmented genome datasets,” said Dr. Corradi.
To know for sure what was going on in AMF heterokaryons, the team needed a method to sequence the complete genomes of both populations of nuclei, allowing more complex studies of gene expression, genetic exchange and evolution in these puzzling fungal packages.
Would you prefer carrots or chicory?
Working with Phase Genomics, Dr. Corradi and his team employed a combination of proximity ligation (Hi-C) and PacBio HiFi data to sequence the genomes of both nuclear populations in four Rhizophagus AMF heterokaryon strains. Surprisingly, all four strains harbored genomes largely similar in structure — 32 chromosomes, with clear delineations between gene-rich and gene-poor regions — but highly divergent in sequence. For all four strains, the two populations of nuclei were essentially haplotypes, derived from parental strains during prior sexual reproduction.
Equipped with eight complete genomes — two haplotypes among four strains — the team followed-up with gene-expression analyses and discovered that each haplotype was transcriptionally active. But within an individual strain, haplotype gene expression patterns were not equal.
“AMF heterokaryons carry two haplotypes that physically separate among many thousands — potentially millions — of co-existing nuclei,” said Dr. Corradi. “This is unheard of in any other organism. But each ‘parental genome’ also regulates different biological functions, and these change depending on the plant host.”
They recorded at times dramatic shifts in haplotype abundance and expression depending on the AMF heterokaryon’s plant host — carrot versus chicory, for example. This suggests that each haplotype makes specific and unique contributions to the AMF heterokaryon’s phenotype. Future studies will have to tease out what role the plant host is playing, if any, in these shifting expression and abundance patterns.
Sex, but when? And more new mysteries
In assembling these long-sought genomes that co-exist within a common cytoplasm, Hi-C has revealed that Rhizophagus AMF heterokaryons are not as complex as once thought, or feared. Both haplotypes within each heterokaryon appear to arise through some past sexual reproduction event, contribute to the AMF’s phenotype and have unique gene expression patterns based on plant host. Their surprisingly ordinary genetic behavior — at least, ordinary for fungi — means it could be possible to engineer AMF that are even better symbionts for specific hosts, helping to boost crop biomass or improve resilience, for example. Engineered strains could also aid in soil remediation, or store carbon that would otherwise end up above ground or in the air.
The findings, coupled with the team’s previous experiments, also bring new mysteries into focus: AMF strains appear to employ a mixture of sexual and asexual reproduction, similar to other fungi. But scientists have never witnessed AMF sexual reproduction — a potentially useful tool for engineering strains. The new genome sequences will also serve as a point of comparison as scientists investigate whether the hundreds of other AMF species are similar to Rhizophagus — and their potential to transform agriculture.







 LinkedIn
LinkedIn Email
Email