Since its debut, next-generation sequencing has not rested on its laurels. Improved sequencing platforms have reduced error and lengthened reads into the tens of thousands of bases. The debut of ultra-long-range sequencing methods that are based on proximity ligation (aka Hi-C) has brought a new order-of-magnitude into reach by linking DNA strands with their neighbors before sequencing.
Rapid progress in this field has birthed genome-resolved metagenomics, the sequencing and assembly of genomes from environmental samples to study ecosystem dynamics. But metagenomic experiments often undersample microbial diversity, missing rare residents, overlooking closely related organisms (like bacterial strains), losing rich genetic data (like viruses and metabolite gene clusters), and ignoring host-viral or host-plasmid interactions.
A revolution within a revolution
New sequencing platforms and methods can reform metagenomics from within. Phase Genomics has been a leader in genome-resolved metagenomics with its ProxiMeta™ platform, which leverages a method that physically connects DNA molecules inside cells before sequencing to generate highly complete genomes for novel bacteria and viruses. Boosting proximity-fueled methods with long-read platforms, such as the PacBio® Sequel® IIe system that can yield HiFi reads of up to 15,000 base pairs with error rates below 1%, could stretch its potential even further.
In a study published in Nature Biotechnology, a team — led by Dr. Timothy Smith and Dr. Derek Bickhart at the U.S. Department of Agriculture and Dr. Pavel Pevzner at the University of California, San Diego — employed both PacBio HiFi sequencing and ProxiMeta in a deep sequencing experiment to uncover record levels of microbial diversity from a fecal sample of a Katahdin lamb. Combined, PacBio HiFi sequencing and ProxiMeta eased assembly, recovered rare microbes, resolved hundreds of strains and their haplotypes, and revealed hundreds of novel plasmid and viral interactions.
Deeper diversity
The team constructed SMRTbell® libraries to generate HiFi data, and ProxiMeta™ libraries to generate long-range sequencing data. The two datasets allowed them to assemble contigs and create draft genomes without manual curation.
Researchers compared the breadth and depth of HiFi data-derived metagenome-assembled genomes, or MAGs, to control MAGs from assemblies of the same sample made using long, more error-prone reads. HiFi data yielded 428 complete MAGs from bacteria and archaea — a record number from a single sample. HiFi data also generated more low-prevalence MAGs, capturing a larger slice of the community’s diversity by picking up more genomes from less common residents.
The hidden actors
But no assembly method could be considered “complete” if it overlooked viruses, the most numerous members of virtually all ecological niches on Earth. These tiny players shape microbial communities in ways scientists are still trying to understand. For example, as agents of horizontal gene transfer, they help spread antibiotic resistance genes. And conversely, they have recently grown in popularity as a means to kill resistant bacteria in our ever-waging war against antibiotic resistance.
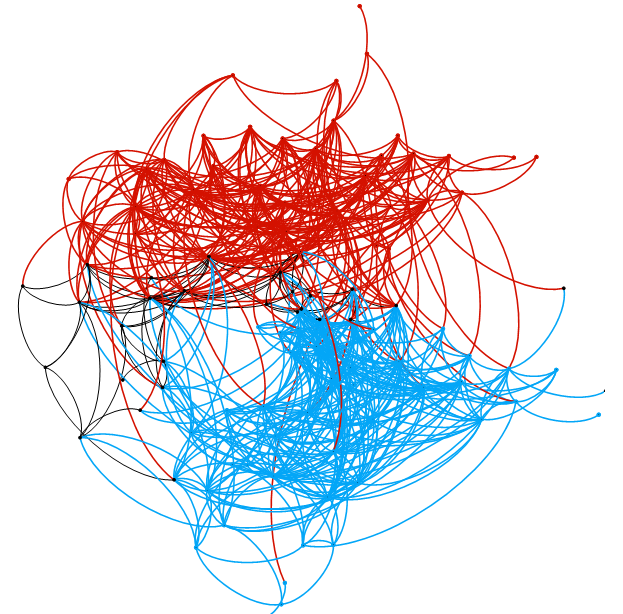
Phase Genomics’ ProxiPhage™ tool can already assemble high-fidelity viral genomes from microbial communities, even using only short-read sequencing data. But the new study shows that having HiFi helps considerably. The team identified 424 unique viral-host interactions, including 60 between viruses and archaea, which is a more than 7-fold increase over control samples. In total, the HiFi library included nearly 400 viral contigs, more than half of which came from a single family that infects bacteria and archaea. The ability to connect viruses with their microbial hosts in vivo is a unique property of Phase Genomics’ technology.
HiFi family trees
The long-range ProxiMeta libraries contained information that yielded more than 1,400 complete and 350 partial sets of gene clusters from archaea and bacteria for synthesizing metabolites such as proteasome inhibitors — the most uncovered to date. These clusters likely help some of these microbes colonize the gut. HiFi data picked up about 40% more clusters than control MAGs, illustrating just how much data is lost when long reads aren’t also highly accurate reads.
The team also used the HiFi-based MAGs to trace lineages within the community. They computationally resolved 220 MAGs into strain haplotypes, based largely on variations within single-copy genes. One MAG had 25 different haplotypes, which are likely strains of the same genus or species.
ProxiMeta ultra-long-range sequencing also linked nearly 300 HiFi-assembled plasmids to specific MAGs — revealing the species that hosted them in vivo. One plasmid, for example, was found in bacteria from 13 different genera. Long-range data also identified the first plasmids associated with three archaea, including Methanobrevibacter and Methanosphaera.
What’s around the bend?
This study has lessons beyond one lamb’s gastrointestinal tract. It shows decisively that the discovery power innate to long-range sequencing methods like ProxiMeta are greatly enhanced when wedded to high-accuracy sequencing methods like HiFi. Together, the two generate increasingly sophisticated metagenome assemblies for biologists to interrogate.
Applied to other environmental samples, this platform could illuminate the diversity and complexity of other microbial communities — from the bottom of the sea to mountain peaks, and within the body of every human being. It could probe pressing issues of our day, such as disease, soil health, and antibiotic resistance, a scourge whose spread and potential solutions — such as phage therapy — can only be forged through a thorough understanding of microbial diversity, interactions, and ecology.


 LinkedIn
LinkedIn Email
Email