
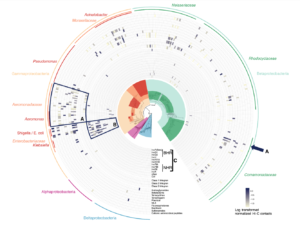
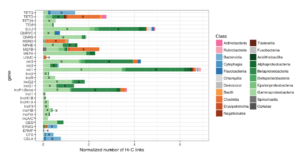
Phase Genomics continues to pioneer genomic innovation, driving advancements in human health and life science research over another impactful year. Our efforts range from developing novel tools for detecting chromosomal abnormalities to managing the world’s most extensive phage-host interaction atlas, thereby accelerating genomic research and promoting a healthier future.
Our ultra-long-range sequencing technology is fostering advancements across a wide array of research applications, both at Phase Genomics and in laboratories worldwide. Utilizing our microbial platform, we are driving transformative discovery in metagenomics and ecology while making bounds in human health with cutting-edge approaches to antimicrobial research and oncology.
Thank you to our supporters, collaborators, and clients for their contributions to making this an outstanding year. Here are some key highlights from 2024.
Insights in oncology

Cow burps, super bugs, and our enemy’s enemy–phages

A novel approach to vaccination

Diving into the data
Two new data analysis tools were made available to ProxiMeta and CytoTerra platform users this year: ProxiMeta™ Explorer and CytoTerra® Curator.
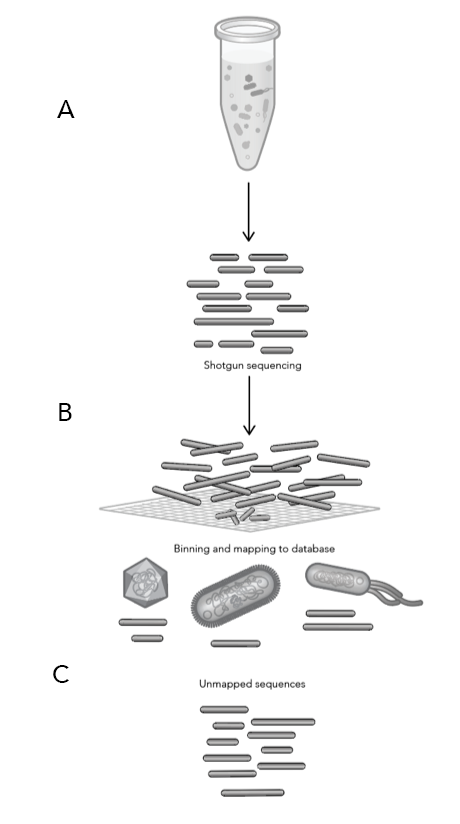
ProxiMeta™ Explorer is an interactive, cloud-based genome-resolved metagenomic analysis platform for data visualization and exploration. The platform provides fully customizable analyses and reports for tracking genomes across time, conditions, groups, and more with a click of a button.
CytoTerra® Curator enables users to effortlessly review, revise, and generate reports from cytogenetic data – no prior bioinformatics experience required. From curating calls to constructing circos plots, Curator provides fast, accurate insights for human genomics and oncology research.
Tune in to this year’s podcasts
Listen to Phase Genomics CEO, Ivan Liachko, discuss the breadth of applications supported by Phase Genomics’ ultra-long-range sequencing technology in these podcast episodes. Discover the story behind commercializing and implementing biotech innovations and get a glimpse at where this technology is taking us.
- Biotech Insights – Biotech CEO Podcast
- Lab Rats to Unicorns – From Genes to Genomes: The Courage to Innovate with Dr. Ivan Liachko
Looking Forward
In 2025, we aim to elevate our technology to new heights and broaden our impact across science and medicine. We hope you will follow us on our journey on X, LinkedIn, and BlueSky as we lead genomics innovation to an insightful and healthy future.
Happy New Year from our team at Phase Genomics!



 It is estimated that ~200 million people worldwide are exposed to arsenic concentrations exceeding current safety standards. Our collaborators have recently demonstrated that mice and human microbiomes can protect mice from arsenic toxicity. While human stool supplementation fully restores protection to arsenic in germ-free mice, researchers were only able to isolate one microbe, Faecalibacterium prausnitzii, that successfully conferred protection to both parent and infant mice. These results are huge because arsenic poses the highest lifetime risk for developing cancer in humans.We will investigate the role of arsenic-transforming bacteria within the gastrointestinal (GI) microbiome as another possible risk factor.
It is estimated that ~200 million people worldwide are exposed to arsenic concentrations exceeding current safety standards. Our collaborators have recently demonstrated that mice and human microbiomes can protect mice from arsenic toxicity. While human stool supplementation fully restores protection to arsenic in germ-free mice, researchers were only able to isolate one microbe, Faecalibacterium prausnitzii, that successfully conferred protection to both parent and infant mice. These results are huge because arsenic poses the highest lifetime risk for developing cancer in humans.We will investigate the role of arsenic-transforming bacteria within the gastrointestinal (GI) microbiome as another possible risk factor. Approximately 13 million rural, urban, and suburban US residents reported owning backyard poultry (BYP) in 2014, and interest in BYP ownership is nearly four times that amount. BYP ownership has risen recently due to product quality, public health, ethical, and animal welfare concerns of commercial operations. However, BYP ownership and disease treatment is largely under-regulated, unlike commercial poultry production. Lack of regulation poses public health concerns of transmission of antimicrobial resistant (AMR) bacteria, such as AMR strains of Salmonella, Mycoplasma gallisepticum, and Escherichia coli commonly associated with BYP. BYP owners (2014 survey) were largely uninformed about poultry diseases and treatments but were interested in learning more on disease management.
Approximately 13 million rural, urban, and suburban US residents reported owning backyard poultry (BYP) in 2014, and interest in BYP ownership is nearly four times that amount. BYP ownership has risen recently due to product quality, public health, ethical, and animal welfare concerns of commercial operations. However, BYP ownership and disease treatment is largely under-regulated, unlike commercial poultry production. Lack of regulation poses public health concerns of transmission of antimicrobial resistant (AMR) bacteria, such as AMR strains of Salmonella, Mycoplasma gallisepticum, and Escherichia coli commonly associated with BYP. BYP owners (2014 survey) were largely uninformed about poultry diseases and treatments but were interested in learning more on disease management. We propose a metagenome characterization of contaminated Munger Landing sediment located in the St. Louis River, Duluth, MN USA. Seasonal samples are already collected and stored; of which one will be sequenced. Munger landing, is located downstream from the U.S. Steel Superfund site and contaminants include PAHs, dioxins, PCBs, and heavy metals.
We propose a metagenome characterization of contaminated Munger Landing sediment located in the St. Louis River, Duluth, MN USA. Seasonal samples are already collected and stored; of which one will be sequenced. Munger landing, is located downstream from the U.S. Steel Superfund site and contaminants include PAHs, dioxins, PCBs, and heavy metals. Hydrothermal vents replenish the oceans with much-needed micronutrients, spewing iron, magnesium, nickel, and other metals from the earth’s crust. These metal micronutrients are used as biological cofactors for organisms throughout the marine food chain. Boiling, sterile hydrothermal fluids quickly cool and are colonized by highly specialized microorganisms that begin to cycle the metal species mixing with the seawater. Though regularly sampled, rarely have hydrothermal plumes been tracked through the water column to establish how microbial colonization occurs through time and space. We lack understanding regarding the replicability of colonization to what extent stochastic processes shape microbial community structure.
Hydrothermal vents replenish the oceans with much-needed micronutrients, spewing iron, magnesium, nickel, and other metals from the earth’s crust. These metal micronutrients are used as biological cofactors for organisms throughout the marine food chain. Boiling, sterile hydrothermal fluids quickly cool and are colonized by highly specialized microorganisms that begin to cycle the metal species mixing with the seawater. Though regularly sampled, rarely have hydrothermal plumes been tracked through the water column to establish how microbial colonization occurs through time and space. We lack understanding regarding the replicability of colonization to what extent stochastic processes shape microbial community structure.












 LinkedIn
LinkedIn Email
Email