SEATTLE (March, 4, 2024) – Phase Genomics, Inc., a leading innovator at the forefront of genomics technology development, today announced $1.5MM in new funding from the Bill & Melinda Gates Foundation to fuel a new antimicrobial discovery platform. Leveraging the power of lysins, phage-derived proteins that selectively kill specific bacteria and archaea, the program aims to address two immediate threats that will shape the next century: a growing global antibiotic resistance crisis and the challenge of reducing global greenhouse gas emissions. The foundation of this effort rests on Phase Genomics’ proprietary global phage atlas, developed with support from the Gates Foundation. Under this project, Phase Genomics will deploy its platform to develop antimicrobial agents that bypass resistance against Campylobacter infections and methanogenic archaea in ruminants that drive global methane emissions.
“Our work at the frontier of microbiome research has unlocked a wealth of new insights on phages, the viruses that infect bacteria. Now, with support from the Gates Foundation, we’re harnessing our global phage database with the goal of improving human and environmental health and providing a critical alternative to traditional antibiotics,” said Ivan Liachko, PhD, founder and CEO of Phase Genomics. “The need for breakthrough therapeutics to combat the growing AMR crisis is urgent. We’ve built the right technology to identify and engineer lysin candidates primed to combat microbes both in environmental settings as well as emerging AMR biothreats and help overcome the industry-wide inertia facing novel antibiotic development.”
Derived from bacteriophage (or simply, phage) genomes, lysins are highly specific lytic proteins that kill bacteria by dismantling the cell wall structure, sparing off-target healthy microbes that are often collateral damage in traditional, systemic antibiotic treatment. Lysin-based antibiotics are well-suited for rapid, scalable biomanufacturing and deployment. Targeted bacteria are also much less likely to develop resistance to lysins than both traditional antibiotics and intact phages, providing a sustainable and durable framework to counter the accelerating antibiotic resistance threat.
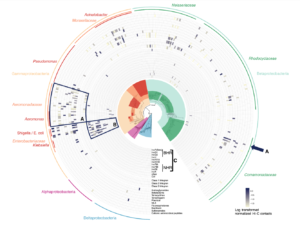
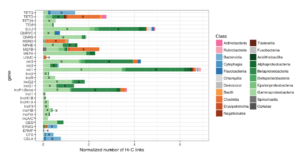
The new platform will build on data from Phase Genomics’ bacteriophage discovery engine which holds one of the world’s largest and most comprehensive collections of phage-microbe interactions containing hundreds of thousands of new host-resolved phage genomes. This continuously-growing phage interactome atlas is primed for the rapid discovery of wide-ranging classes of antimicrobial lysins derived from phages. The platform is superior to other approaches in both scale and accuracy, simultaneously resolving both microbial targets and the phages that infect them, with each pair containing a potential target-specific lysin candidate. Phase Genomics’ ProxiMeta™-powered phage atlas forms a deep well of target bacterial pathogens and new candidate biologics to tackle emerging drug-resistant pathogens and environmental biothreats.
This year-long project also marks a first-of-its-kind collaboration between Phase Genomics and Seattle-based Lumen Bioscience, who will assess lysin bioactivity in their robust and scalable microbial expression system.
Follow Phase Genomics on X and LinkedIn for the latest news and information.
About Phase Genomics
Phase Genomics applies proprietary proximity ligation technology to enable chromosome-scale genome assembly, microbiome discovery, as well as analysis of genomic variation and genome architecture. In addition to a comprehensive portfolio of laboratory and computational services and products, including kits for plants, animals, microbes, and human samples, they also offer an industry-leading genome and metagenome assembly and analysis software.
Based in Seattle, WA, the company was founded in 2015 by a team of genome scientists, software engineers, and entrepreneurs. The company’s mission is to empower scientists with genomic tools that accelerate breakthrough discoveries.
Contact
Eric Schudiske
eric@s2spr.com


 LinkedIn
LinkedIn Email
Email