
Decades of antibiotic use – and abuse – are triggering a global rise in antibiotic resistance and limiting the usefulness of these life-saving drugs. In a nod to the adage, “The enemy of my enemy is my friend,” a solution may lie with bacteria’s oldest adversary: phages, the viruses that prey upon them. Our team at Phase Genomics is harnessing groundbreaking new metagenomic data and AI to tap into the evolutionary innovations of phages – and to eradicate dangerous microbial pathogens with surgical precision.
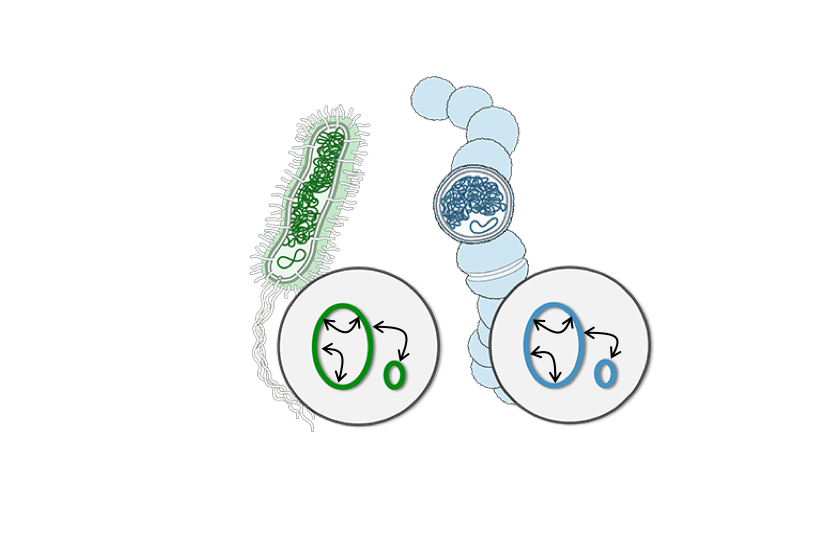
The need could not be greater. Fewer new antibiotics are hitting the market. The UN estimates that by 2050, worldwide deaths from antibiotic-resistant “superbugs” will overtake deaths from cancer. Early 20th century scientists explored deploying phages to cure bacterial infections, an idea that has been recently resurrected. Phages are a staggeringly diverse class of bacteria-killers. By one estimate there are 1031 of them on this planet right now, vastly more than all living organisms combined. But using phages to cure infections has its own drawbacks: Mass production is difficult since phages only grow in bacteria, which can be difficult to culture, and it turns out bacteria have a barrage of defenses against intact viruses, imparting resistance against them.
While phages present one opportunity to help us stave off a return to the pre-penicillin past, we can also use their anti-bacterial weapons to launch a new arsenal rooted in synthetic biology. Phages produce proteins called lysins to destroy their hosts’ cell walls. These proteins have evolved over millennia to specifically target the phages’ hosts. They can be purified and used as precision antimicrobials, molecules that specifically kill the target bacteria without the collateral damage and resistance brought about by traditional wide-spectrum antibiotics.
Our team has used our unique genome sequencing technology to build the world’s largest catalog of the genomes of phages and the microbes that they attack – including the sequences of lysin proteins that they make. We’re harnessing this catalog to design, synthesize, and perfect lysin-based therapeutics that can attack bacterial pathogens safely, effectively, and with a surgical precision that today’s antibiotics lack.
Lysins hold tremendous advantages over traditional antibiotics. Antibiotics take out swathes of bacteria in our microbiomes that are essential for good health, leaving us more vulnerable to future infections – like the dreaded C. difficile – as well as to immune dysregulation. Yet most lysins target only the phage’s host species and its close relatives. And though antibiotic resistance spreads rapidly via plasmids, bacteria struggle to evolve resistance to exogenously introduced lysins.
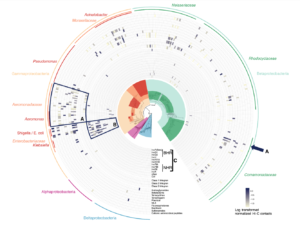
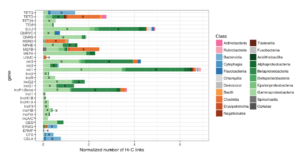
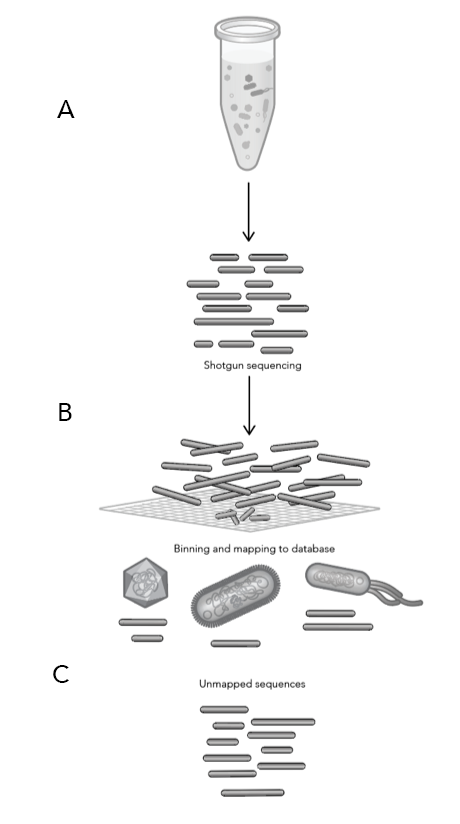
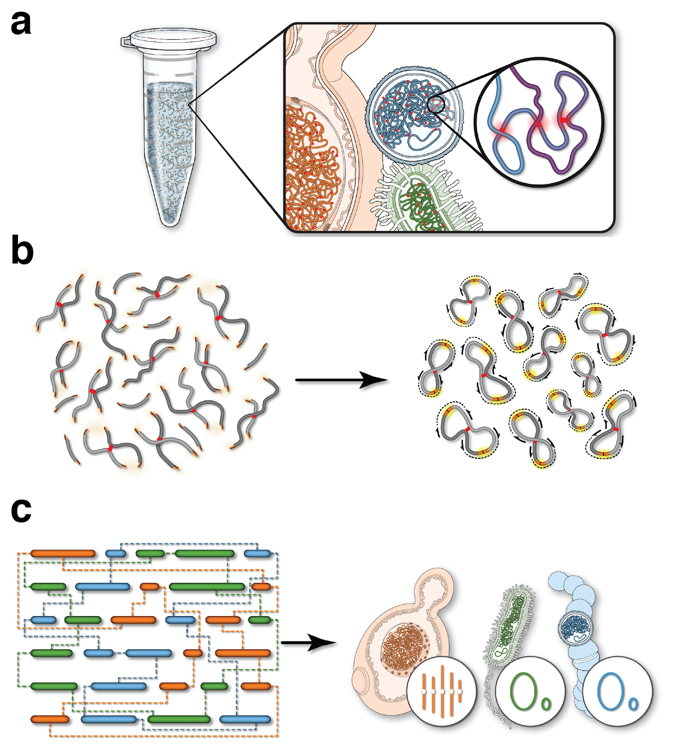
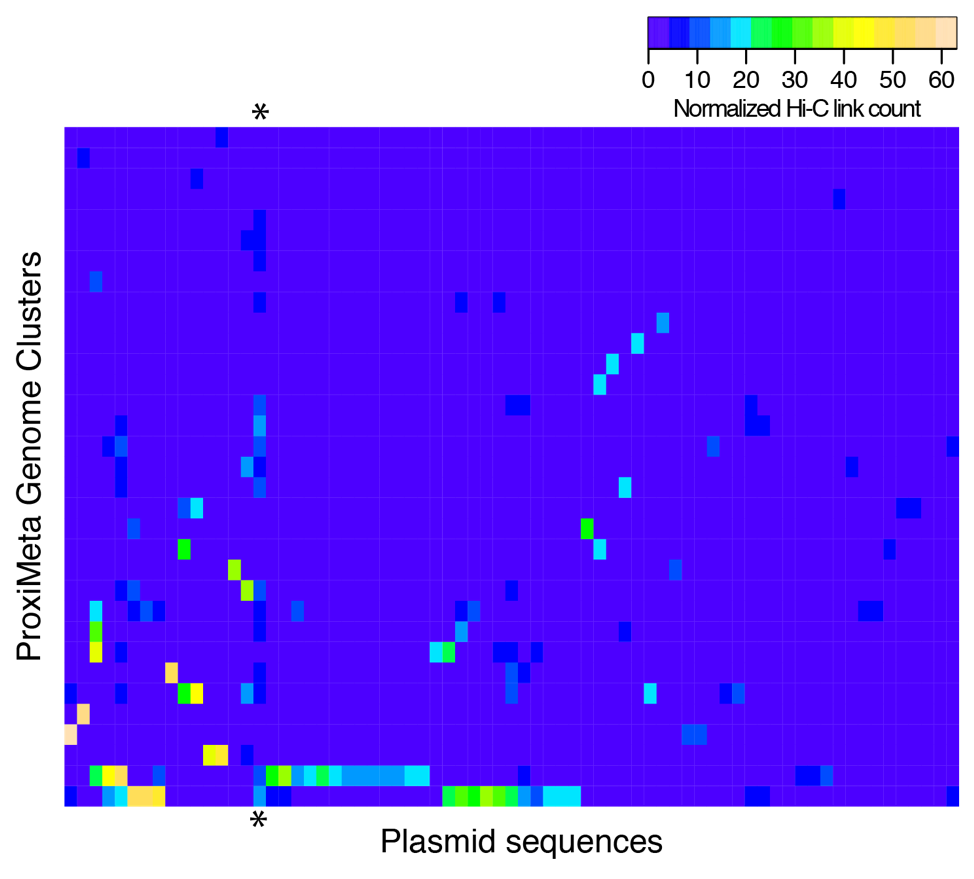
Our collective knowledge of lysins to date comes largely from isolated experiments on phages or small-scale genomic studies. To deploy lysins as a life-saving solution, we need detailed knowledge of the intricate and intimate interactions between phages and bacteria. Phase Genomics has led this effort by building a vast catalog containing hundreds of thousands of phage genomes from different microbial environments. Our proprietary ProxiMeta technology employed for these experiments preserves unique information about essential ecological interactions in these microbiomes, including the host bacterial species that specific phages target. Thanks to this large and growing catalog of phage-microbe interactions, for many pathogenic bacteria, we can find specific lysins that could turn its cell walls into Swiss cheese.
We are using this foundational knowledge to build the first foundry for lysins. With support from the Bill and Melinda Gates Foundation, Phase Genomics is collaborating with Lumen Bioscience to design, grow, and purify lysins identified by our catalog. This proving ground will serve as the foundation for a future pipeline for lysin design – augmented by machine learning to hone target specificity, perfect performance and even create entirely new lysins with a desired target specificity. To make a custom-designed lysin against almost any bacteria, we would need to find a phage – and its lysin – that attacks it. This approach to lysin research and discovery has applications even beyond medicine, such as critically needed environmental remediation.
Our goal to develop therapeutic lysins would upend the existing paradigm for treating bacterial infections. Today, medical professionals have a shrinking pool of imperfect antibiotics that cut a swathe through our microbiomes to take out the bacterial bad guys. With lysins on the shelf as an option, we would be taking away this machete, and replacing it with a scalpel.

















 LinkedIn
LinkedIn Email
Email