
Hydrothermal mat sampling aboard R/V Roger Revelle using ROV Jason. Credit: R/V Roger Revelle, Scripps institute of Oceanography.
For decades, biologists largely studied microbes and their viruses in isolation, nurtured in laboratory cultures. Yet, to paraphrase the poet John Donne, no microbe is an island. In recent years, scientists have recognized this by studying microbes not as individual species, but as part of the larger microbiome: the communal ecosystems, each home to many different types of bacteria and archaea, in which most microbes reside. It is in these realms that microbes display their collective might. From guts to geysers, tiny tales of competition and cooperation within microbiomes have big effects on our health and environment — such as the spread of antibiotic resistance and the stability of food webs.
Revealing microbiome mechanics
Traditional, laboratory-based methods struggle to probe the individual components of the microbiome. But “metagenomics” allows us to study the community at large. Metagenomics is the sequencing of DNA from microbial communities, and metagenome-assembled genomes — or MAGs — put together using ever-more sensitive tools and processes, are increasingly able to resolve the inner workings of these complex ecosystems.
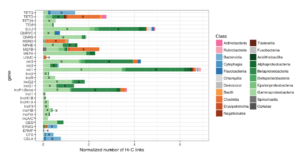
Recently, a collaboration between Phase Genomics and a team at Harvard University on a metagenomics project showed that phages — viruses that infect bacteria and archaea — have a surprisingly broad impact on the microbiome of a seafloor hydrothermal vent. Using a technique called proximity ligation (Hi-C), which cross-links DNA strands from the same cell before DNA extraction and sequencing, researchers reconstructed MAGs in this community and found that diverse microbes, including bacteria and archaea separated by billions of years of evolution, sported records of past encounters with the same phages. One explanation is that the phages have an unheard-of level of host diversity — one certainly not predicted by laboratory experiments. Another is that these deep-sea microbes may somehow “share” adaptive immunity across broad and deep evolutionary gulfs.
If phages have similarly broad impacts far above the ocean floor, scientists may have to rethink how communication, cooperation and evolution shape microbiomes — and how they impact the larger creatures, like us, that depend on them.
Tapping the archive
Microbiomes teem with phages. But deciphering their reach is no easy task. Thankfully, some bacteria and archaea are hoarders. Their CRISPR-based immune responses record past phage infections by inserting short fragments of phage genomes into a specific region of their own genome. Some studies have even sought to reconstruct the reach of phages in a microbiome by probing the content of these areas — known as spacer regions. Yet, the approach has its drawbacks.
“Spacer regions are rich in repeats, so they don’t get sorted well in the MAG assembly process,” said Yunha Hwang, a doctoral student at Harvard University. “That creates a bias regarding which spacers and phage fragments are ultimately assembled into MAGs.”
Hwang has studied these genetic archives of microbial immunity, and previously reported that, in a desert microbiome, phages may have broad host ranges.
“It was a preliminary result, but very exciting,” said Hwang. “I wanted to see if this was a wider feature of microbiomes, and I wanted to avoid that assembly bias.”
Achieving Hi-C depth in deep oceans
Hwang and Peter Girguis, a professor at Harvard, worked with Phase Genomics to employ a metagenomic approach centered on Hi-C, which, by preserving physical linkages between DNA fragments present in the same cell, eases the process of resolving repeat-rich regions like CRISPR spacers.
Hwang collected samples from the microbiome near a hydrothermal vent in the Gulf of California’s Guyamas Basin. Microbial communities like this employ “alternative” metabolic pathways — relying on the plume’s rich geochemical outflow for nutrients, energy and raw materials instead of the sun-based food webs more familiar to surface-dwellers. As soon as she reached port in San Diego, Hwang shipped the microbiome samples to Phase Genomics for cross-linking, DNA extraction, sequencing and MAG assembly.
The spacer regions of the MAGs assembled via Hi-C showed similar profiles of past phage infection compared to conventional spacer-sequencing and assembly. But the higher-quality Hi-C MAGs also eased the search for phage fragments within CRISPR spacers. And, as in Hwang’s study of desert microbiomes, individual phages in the hydrothermal vent microbiome had a broad reach — including bacteria to archaea.
“This was so baffling to us, because these are two separate domains of life,” said Hwang. “The ability for a phage to infect a host depends on fundamental properties of cell biology, and bacteria and archaea are so different — their membranes, their proteins, their genomes. So, what does this mean?”
Another puzzle is that bacteria and archaea that are linked by symbiotic relationships — such as eating one another’s metabolic leftovers — were also more likely to harbor genomic fragments of the same phages in their CRISPR spacers.
Spread the word
One theory to explain these findings is that phages within microbiomes, which can be hard-pressed for space in these close-knit communities, have evolved to infect hosts with radically diverse membrane compositions, host defenses and cell biology. But that is not the only possibility. Another is that symbiotic partners, separated by billions of years of evolution but united at the dinner table, may be sharing more than just a meal.
“In symbiotic microbes, when one population or species gets infected by a phage, there could be a selective advantage in sharing that adaptive, genetically encoded immunity with your partners,” said Hwang.
Future metagenomic studies of other microbiomes may help resolve these theories, or sire new ones. But the eventual explanations will undoubtedly force scientists to rethink how genetic information flows within microbiomes.
“How do bacteria and archaea build up ‘resilience’ in such closely packed communities?” said Hwang. “Perhaps one way that happens through selective pressure to share records of past phage infections widely. Keeping your neighbor healthy keeps you healthy.”
Sounds familiar
Once upon a time, far above the ocean floor, children played a game called “telephone”: passing a phrase from one person to another — in the form of a whisper — to see how the message changed as it is heard by each ear and transmitted by each voice.
It seems that bacteria, archaea and phages play similar games, which is just the latest surprise that metagenomics has revealed about microbiomes. It will certainly not be the last.
Pass it on.







 LinkedIn
LinkedIn Email
Email